By Marcello Cherchi, MD PhD
For patients
Wilson’s disease, also called hepatolenticular degeneration, is a genetic disease that lets copper build up in the liver, brain and other parts of the body. The liver problems can cause the skin and eyes to become yellow. The brain problems can cause abnormal movements. If your doctor suspects Wilson’s disease, they may check blood tests, urine tests, brain imaging, a liver biopsy or genetic tests. Treatment is with drugs that take copper out of the body.
For clinicians
Overview
Wilson’s disease (hepatolenticular degeneration) is a genetic disorder of copper metabolism wherein copper accumulates in the liver, brain and other organs. Initial manifestations of the liver disease include jaundice and cirrhosis. Neurological manifestations may include dysarthria, cerebellar ataxia, dystonia, tremor, parkinsonism, choreoathetosis, cognitive impairment and myoclonus. Most patients with neurological symptoms, and half of the patients without neurological symptoms, exhibit copper accumulation at the corneal periphery observable as Kaiser-Fleischer rings. Most patients with neurological Wilson’s disease also exhibit abnormalities in ocular motor function, including abnormalities of vertical (more than horizontal) smooth pursuit, optokinetic responses and saccades. Clues to diagnosis come from physical examination, laboratory findings (e.g., elevated serum ceruloplasmin, elevated urinary copper excretion), brain imaging and liver biopsy (elevated copper concentration); definitive diagnosis comes from genetic testing.
Introduction

Dr. Samuel Alexander Kinnier Wilson (Cedarville, New Jersey, USA 1878 – London, England, 1937) published a detailed report and analysis of 12 cases of hepatolenticular degeneration (Wilson 1912), a disease which eventually bore his name. In his article he summarized (page 436):
“Progressive lenticular degeneration may be defined as a disease which occurs in young people, which is often familial but not congenital or hereditary; it is essentially and chiefly a disease of the extrapyramidal motor system, and is characterised by involuntary movements, usually of the nature of tremor, dysarthria, dysphagia, muscular weakness, spasticity, and contractures with progressive emaciation; with these may be associated emotionalism and certain symptoms of a mental nature. It is progressive, and, after a longer or shorter period, fatal. Pathologically it is characterised predominantly by bilateral degeneration of the lenticular nucleus, and in addition cirrhosis of the liver is constantly found, the latter morbid condition rarely, if ever, giving rise to symptoms during the life of the patient” (Wilson 1912).
At 214 pages, Wilson’s article was the longest ever published in the journal Brain (Compston 2009).
Epidemiology
Reilly and colleagues (Reilly, Daly, Hutchinson 1993) reported that the incidence of Wilson’s disease in Ireland was 17 per 1 million. A literature review by Sandahl and colleagues (Sandahl et al. 2020) found the prevalence of Wilson’s disease to range from 1 in 30,000 to 1 in 40,000.
Genetics
Wilson’s disease (OMIM 277900) is now known to be an autosomal recessively transmitted defect in the ATP7B gene (OMIM 606882) on chromosome 13q14.3 that encodes for adenosine triphosphatase responsible for the excretion of excess copper. The defect permits accumulation of the excess copper predominantly in the liver cornea and brain (basal ganglia, thalamus, midbrain, cerebellum, prefrontal cortex). Less commonly affected organs include the kidney, heart, skin, endocrine system and musculoskeletal system.
Pathophysiological mechanism of disease
As far as the ocular motor abnormalities in Wilson’s disease are concerned, Ingster-Moati and colleagues state that, “The extrapyramidal fibres originating from the striated nucleus and the pallidum play a role of relay in the Cajal and Darkschewitsch nuclei, providing afferent fibres to the ocular motor nuclei through the medial longitudinal fasciculus” (Ingster-Moati et al. 2007).
Clinical presentation
Lesniak and colleagues (Lesniak, Czlonkowska, Seniow 2008) reported that in about 40% of patients the initial symptoms are related to hepatic dysfunction; whereas in about 15% of patients the initial symptoms are neuropsychiatric, including psychosis, antisocial behavior, irritability, impulsiveness, mood disorders, and frontal executive dysfunction.
The copper deposition in the deep epithelial layers at the outer border of the cornea (Descemet’s membrane) can be observed as brownish, grayish or greenish rings; these can be found in about 90% of patients with neurological symptoms, and 50% of patients without neurological symptoms. This ocular finding was described by the German ophthalmologists, Bernhard Kayser (Kayser 1902) and Bruno Fleischer (Fleischer 1903), and came to be known as Kayser-Fleischer rings (Pearce 2019). Kaiser-Fleischer rings can sometimes be seen simply on face-to-face inspection, though are more easily observed on a slit lamp examination. Kaiser-Fleischer rings can also be seen on anterior segment optical coherence tomography (Broniek-Kowalik et al. 2019).
For a time it was thought that Kayser-Fleischer rings were pathognomonic for Wilson’s disease, but it was eventually realized that these can also be found in neonatal cholestasis (Dunn, Annable, Kliegman 1987), other liver diseases, and conditions that result in copper overload.
Physical examination
In Wilson’s disease patients with neurological involvement, physical examination findings may include dysarthria, cerebellar ataxia, dystonia, tremor, parkinsonism, choreoathetosis, cognitive impairment and myoclonus (Lorincz 2010).
Ocular motor examination
Although ocular motor abnormalities are quite common in Wilson’s disease (Ingster-Moati et al. 2007), Wilson’s original description (Wilson 1912) only commented on this in 2 out of the 12 cases he described (Hyman and Phuapradit 1979). Some of these may be difficult to observe on face-to-face examination, and only become apparent on instrumented ocular motor testing.
Testing: vestibular
A rather broad variety of ocular motor abnormalities has been reported in Wilson’s disease, including:
- Slow vertical saccades (Ingster-Moati et al. 2007; Kirkham and Kamin 1974)
- Abnormal vertical smooth pursuit (Ingster-Moati et al. 2007)
- Abnormal horizontal and vertical optokinetic responses (Ingster-Moati et al. 2007)
- Gaze distractibility (Lennox and Jones 1989)
- Oculogyric crisis (Lee, Kim, Lyoo 1999)
- Lid opening apraxia (Keane 1988)
Ingster-Moati and colleagues (Ingster-Moati et al. 2007) studied 34 Wilson’s disease patients using electro-oculography. They reported that 31 (91%) of patients had one or more ocular motor abnormalities, with vertical eye movements affected more than horizontal eye movements. Of all 34 patients:
- 29/34 (85%) had abnormalities of vertical pursuit
- 14/34 (41%) had abnormalities of vertical optokinetic nystagmus
- 14/34 (41%) had abnormalities of horizontal pursuit
- 10/34 (30%) had abnormalities of vertical saccades
- 9/34 (27%) had abnormalities of horizontal optokinetic nystagmus
- 5/34 (15%) had abnormalities of horizontal saccades
- 3/34 (9%) had abnormalities of visual fixation
Given this variety of abnormalities, it is understandable that patients have difficulty reading (Hyman and Phuapradit 1979).
Testing: other
Findings that increase suspicion for Wilson’s disease include the presence of Kaiser-Fleischer rings, elevated serum ceruloplasmin, elevated urinary copper excretion, or elevated copper concentration on liver biopsy. Diagnosis can be confirmed with genetic testing, for which medical genetics consultation is recommended.
Imaging
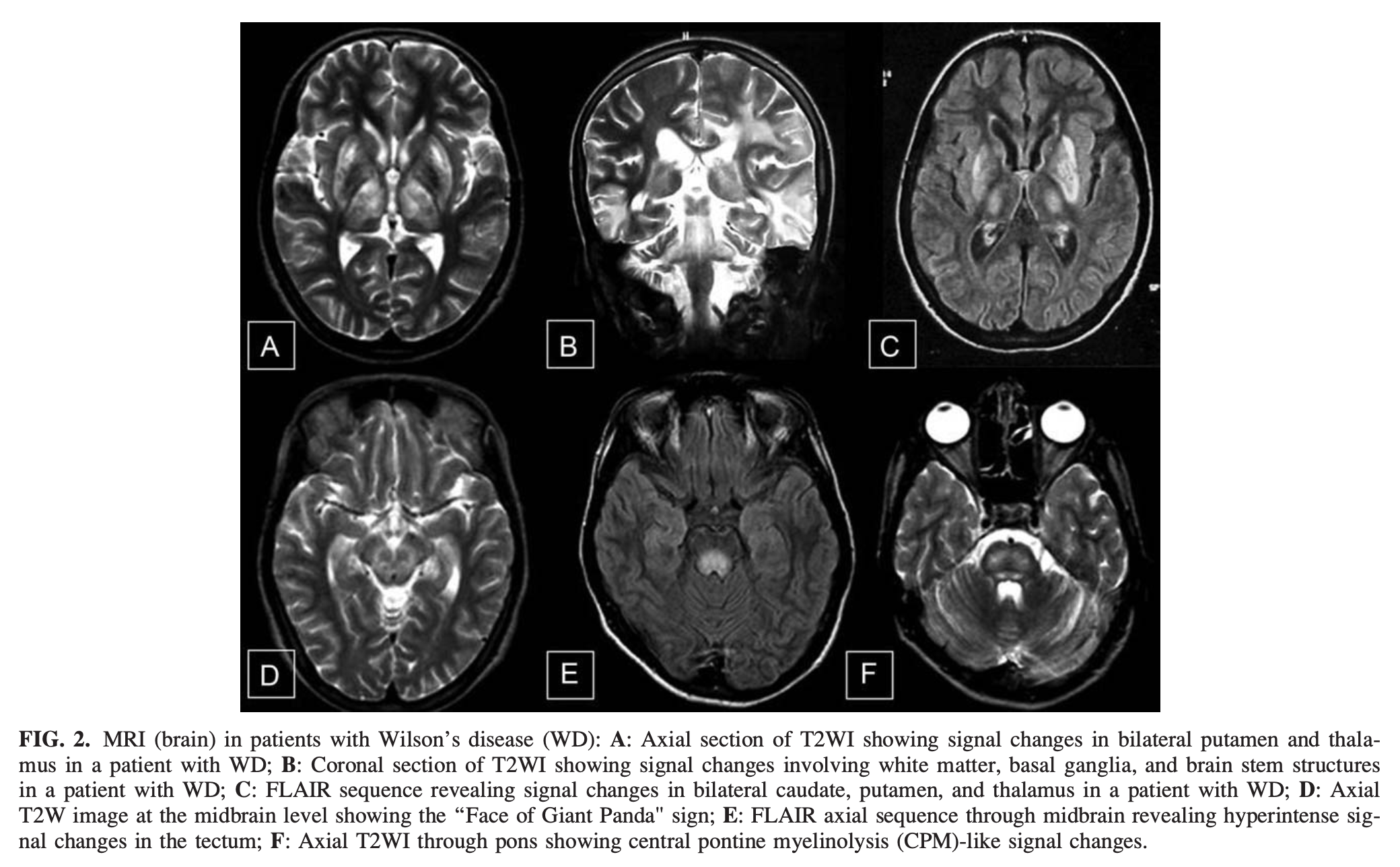
Prashanth and colleagues (Prashanth et al. 2010) retrospectively reviewed 56 patients with Wilson’s disease and observed that:
- 75% had tectal plate hyperintensity
- 63% had central pontine myelinolysis-like abnormalities
- 55% had signal changes in the basal ganglia, thalamus and brainstem
- 14% of cases had the “face of giant panda” sign
They concluded that, “Besides ‘Face of giant panda’ sign, hyperintensities in tectal-plate and central pons (CPM-like), and simultaneous involvement of basal ganglia, thalamus, and brainstem are virtually pathognomonic of WD [Wilson’s disease]” (Prashanth et al. 2010).
The Figure below, from Prashanth and colleagues (Prashanth et al. 2010), shows these imaging findings.
A number of abnormalities are detectable on functional imaging in patients with Wilson’s disease (Tinaz et al. 2021), but this is not part of the standard diagnostic workup.
Histopathology
Liver biopsy shows cirrhosis and elevated copper concentration.
Differential diagnosis
The differential diagnosis of Wilson’s disease includes other basal ganglionic disorders.
Treatment and prognosis
In the United States the most common treatment for Wilson’s disease is the chelating agent D‑penicillamine. Less commonly used agents include, trientene hydrochloride, trientene tetrahydrochloride, ammonium tetrathiomolybdate, or sodium dimercaptosuccinate. Zinc supplementation (e.g., zinc acetate) is sometimes also used for maintenance therapy, because zinc competes with copper absorption. Treatment can decelerate and, in some cases, partially reverse disease progression.
References
Broniek-Kowalik K, Dziezyc K, Litwin T, Czlonkowska A, Szaflik JP (2019) Anterior segment optical coherence tomography (AS-OCT) as a new method of detecting copper deposits forming the Kayser-Fleischer ring in patients with Wilson disease. Acta Ophthalmol 97: e757-e760. doi: 10.1111/aos.14009
Compston A (2009) Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295-509. Brain 132: 1997-2001. doi: 10.1093/brain/awp193
Dunn LL, Annable WL, Kliegman RM (1987) Pigmented corneal rings in neonates with liver disease. J Pediatr 110: 771-6. doi: 10.1016/s0022-3476(87)80022-7
Fleischer B (1903) Zwei weitere Falle von grunlicher Verfarbung der Kornea [Two more cases of greenish discoloration of the cornea]. Klinische Monatsblätter für Augenheilkunde 41: 489-491.
Hyman NM, Phuapradit P (1979) Reading difficulty as a presenting symptom in Wilson’s disease. J Neurol Neurosurg Psychiatry 42: 478-80. doi: 10.1136/jnnp.42.5.478
Ingster-Moati I, Bui Quoc E, Pless M, Djomby R, Orssaud C, Guichard JP, Woimant F (2007) Ocular motility and Wilson’s disease: a study on 34 patients. J Neurol Neurosurg Psychiatry 78: 1199-201. doi: 10.1136/jnnp.2006.108415
Kayser B (1902) Über einen Fall von angeborener grünlicher Verfärbung der Cornea [On a case of congenital greenish discoloration of the cornea]. Klinische Monatsblätter für Augenheilkunde 40: 22-25.
Keane JR (1988) Lid-opening apraxia in Wilson’s disease. J Clin Neuroophthalmol 8: 31-3. doi: 10.3109/01658108808996020
Kirkham TH, Kamin DF (1974) Slow saccadic eye movements in Wilson’s disease. J Neurol Neurosurg Psychiatry 37: 191-4. doi: 10.1136/jnnp.37.2.191
Lee MS, Kim YD, Lyoo CH (1999) Oculogyric crisis as an initial manifestation of Wilson’s disease. Neurology 52: 1714-5. doi: 10.1212/wnl.52.8.1714
Lennox G, Jones R (1989) Gaze distractibility in Wilson’s disease. Ann Neurol 25: 415-7. doi: 10.1002/ana.410250417
Lesniak M, Czlonkowska A, Seniow J (2008) Abnormal antisaccades and smooth pursuit eye movements in patients with Wilson’s disease. Mov Disord 23: 2067-73. doi: 10.1002/mds.22276
Lorincz MT (2010) Neurologic Wilson’s disease. Ann N Y Acad Sci 1184: 173-87. doi: 10.1111/j.1749-6632.2009.05109.x
Pearce JMS (2019) Historical note: the Kayser-Fleischer ring. Advances in Clinical Neuroscience and Rehabilitation. doi: 10.47795/XCRJ9055
Prashanth LK, Sinha S, Taly AB, Vasudev MK (2010) Do MRI features distinguish Wilson’s disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Mov Disord 25: 672-8. doi: 10.1002/mds.22689
Reilly M, Daly L, Hutchinson M (1993) An epidemiological study of Wilson’s disease in the Republic of Ireland. J Neurol Neurosurg Psychiatry 56: 298-300. doi: 10.1136/jnnp.56.3.298
Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P (2020) The Prevalence of Wilson’s Disease: An Update. Hepatology 71: 722-732. doi: 10.1002/hep.30911
Tinaz S, Arora J, Nalamada K, Vives-Rodriguez A, Sezgin M, Robakis D, Patel A, Constable RT, Schilsky ML (2021) Structural and functional brain changes in hepatic and neurological Wilson disease. Brain Imaging Behav 15: 2269-2282. doi: 10.1007/s11682-020-00420-5
Wilson SAK (1912) Progressive lenticular degeneration: a familial nervous dysease associated with cirrhosis of the liver. Brain 34: 295-507. doi: 10.1093/brain/34.4.295
![]()