By Marcello Cherchi, MD PhD
For patients
Episodic ataxias (EA) are a set of inherited disorders that cause episodes of disequilibrium, clumsiness and sometimes other symptoms, lasting minutes to hours, sometimes triggered by stress, by physical exertion or by dietary factors. Episodes often begin in childhood, and may occur a few times per year or multiple times per day. Patients feel normal (or mostly normal) in between episodes. Some patients improve with the oral medication acetazolamide. If acetazolamide fails, then your doctor may consider referring you to a specialist in ataxia, or to a genetics counselor.
For clinicians
Overview
Episodic ataxias (EA), and more broadly the so-called acetazolamide-responsive ataxias, are due to genetic defects in ion channels. Most of these diseases are familial, inherited in an autosomal dominant fashion. Symptoms often, though not always, begin in childhood. EAs are rare, but among them, EA1 and EA2 were the first described, and are the most common. EA3-6 are much rarer. In many cases episodes are triggered by “stress” (emotional duress), physical exertion or dietary factors (e.g., caffeine). The frequency of the episodes is extremely variable. The duration is also variable, though generally, the episodes in EA1 last minutes, while those in EA2 last hours. Typical symptoms include disequilibrium, ataxia/incoordination and dysarthria; less common symptoms include visual symptoms, weakness, tremor, headache and nausea. EA1 and EA2 have been connected to a broad range (dozens each) of genetic mutations, though the mechanism by which these mutations provoke neurological dysfunction is unclear. During an episode patients with EA1 and EA2 may exhibit a variety of eye movement abnormalities. Inter-ictally some patients exhibit subtle eye movement abnormalities as well. Most of these eye movement abnormalities broadly localize to the cerebellum, but none is specific or sensitive for these disorders. For most of these disorders, many patients will exhibit some improvement with acetazolamide; EA2 patients tend to be most responsive. Several other medications have been tried in the treatment of episodic ataxias, though none has been studied as well as acetazolamide.
In a patient complaining of episodes disequilibrium and incoordination lasting minutes to hours, sometimes triggered by stress, by exertion or by dietary factors, who feels normal (or nearly so) inter-ictally, whose examination is unremarkable, and who has a family history of similar symptoms, EA is a reasonable consideration. Because EAs are rare, the clinician should keep a broader differential diagnosis that includes much more common entities, such as migraine associated vertigo. Imaging (usually an MRI of the brain) is reasonable to exclude structural lesions. A trial of acetazolamide (absent any contraindications) is reasonable. If that is unsuccessful, then referral to a specialist in ataxia, or a genetics counselor, is reasonable.
Introduction
Some sources (Bain, O’Brien et al. 1992, Klaas, Burkholder et al. 2013) credit Harry Lee Parker (1894 – 1959) with the original description of a cohort of patients with what eventually became known as familial episodic ataxia (Parker 1946).

Familial episodic ataxias (EA) are channelopathies manifesting with episodic ataxia and disequilibrium (Baloh and Jen 2002). Episodic ataxia type 1 (EA1) is caused by missense mutations in the potassium channel gene KCNA1 on chromosome 12p13. Episodic ataxia type 2 (EA2) is caused by missense and nonsense mutations in the calcium channel gene CACNA1A on chromosome 19p.
Episodic ataxias type 1 and 2 are quite rare but have been relatively well described. More recently reported episodic ataxias (types 3, 4, 5 and 6) are even more rare, and are still being studied and debated.
These diseases are often grouped together because they share some features, yet there is probably no single feature that unifies them all. They are probably all due to genetic mutations, though for some variants the culprit gene has not yet been identified. They are mostly hereditary, although EA6 has only been identified in a single individual (perhaps as a de novo mutation), so the term “familial” is probably inappropriate. Patients experience episodic symptoms, but inter-ictally may experience milder chronic symptoms at baseline. Most patients exhibit ataxia, but not all. Most types have at least some patients whose symptoms improve with acetazolamide, but not all (such as EA4).
Episodic ataxia type 1
Introduction
In 1975 VanDyke and colleagues described a kindred in which 11 people over 3 generations exhibited episodic ataxia and continuous myokymia (VanDyke, Griggs et al. 1975). Subsequent reports (Hanson, Martinez et al. 1977) identified similar findings in other families. In 1994 linkage analysis identified mutations at a locus on chromosome 12p13 (Litt, Kramer et al. 1994) that was identified as a gene that encodes the KCNA1 potassium channel (Browne, Gancher et al. 1994). This eventually led to this being designated as a familial episodic ataxia, and it was ultimately called “episodic ataxia type 1,” when other types were discovered.
Episodic ataxia type 1, various genetic mutations
EA1 is inherited in an autosomal dominant fashion.
A broad range of genetic mutations has been described in EA1 (Bretschneider, Wrisch et al. 1999, Eunson, Rea et al. 2000, Knight, Storey et al. 2000, Klein, Boltshauser et al. 2004, Imbrici, Gualandi et al. 2008, Shook, Mamsa et al. 2008, D’Adamo, Gallenmuller et al. 2014, Lassche, Lainez et al. 2014, Petitjean, Kalstrup et al. 2015, Chen, Fu et al. 2016, Mestre, Manole et al. 2016, Karalok, Megaro et al. 2018, Yuan, Yuan et al. 2020, Lee, Kim et al. 2021).
For each mutation there have been various proposals for the mechanism by which is causes neurological dysfunction (D’Adamo, Liu et al. 1998, D’Adamo, Imbrici et al. 1999, Manganas, Akhtar et al. 2001, Maylie, Bissonnette et al. 2002, Klein, Boltshauser et al. 2004, Imbrici, D’Adamo et al. 2006, Heeroma, Henneberger et al. 2009, Tomlinson, Tan et al. 2010, Imbrici, D’Adamo et al. 2011, Peters, Werry et al. 2011, Zhu, Alsaber et al. 2012, Ferrick-Kiddie, Rosenthal et al. 2017, Zhao, Petitjean et al. 2020).
Episodic ataxia type 1, clinical presentation
Episodes of EA1 can begin in early childhood or adolescence (Kotagal 2012).
Some episodes of EA1 have no apparent trigger. In some patients the triggers are relatively consistent. Graves and colleagues studied 39 patients (51% male), age 15 – 65 years (median 37 years), with genetically confirmed EA1, and reported the triggers listed in the Table below from Graves and colleagues (Graves, Cha et al. 2014).
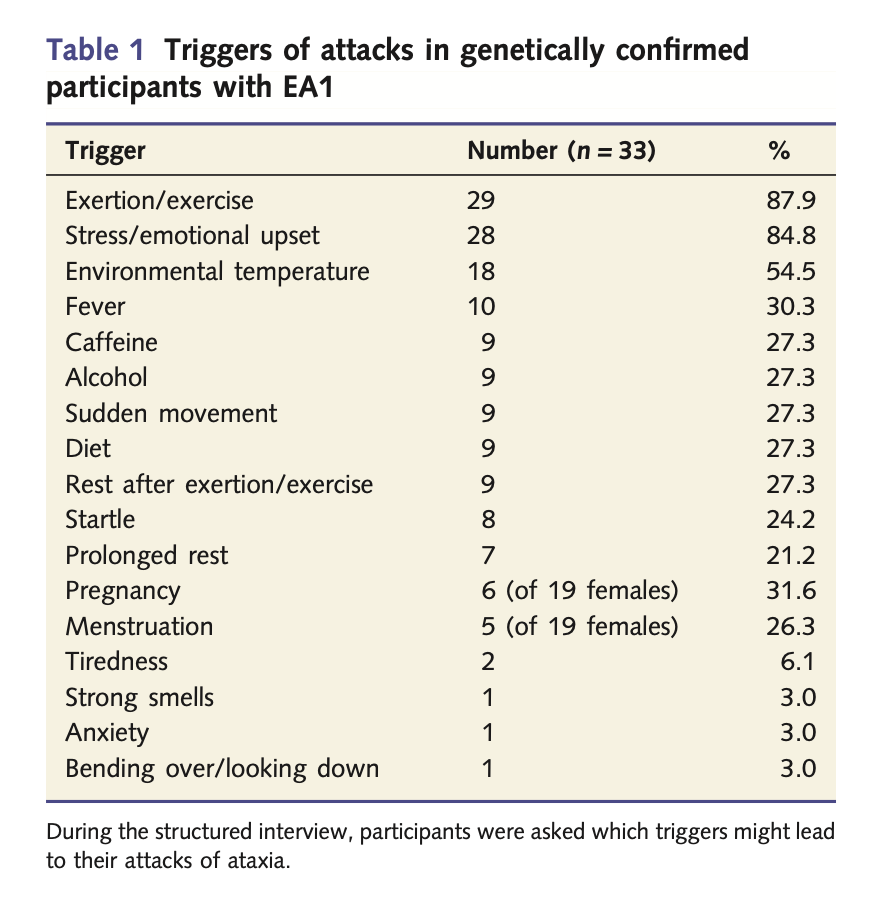
EA1 attacks usually last minutes (Baloh and Jen 2002, Dressler and Benecke 2005), less commonly hours (Graves, Cha et al. 2014). In some patients the episodes can occur with impressive frequency, up to dozens of times per day (Kotagal 2012). Symptoms usually include midline cerebellar dysfunction (truncal ataxia), limb ataxia, dysarthria, dystonia (Dressler and Benecke 2005), and sometimes visual symptoms such as oscillopsia and visual blurring (Rajakulendran, Schorge et al. 2007). The Table below lists the symptoms reported by the patients studied in the series by Graves and colleagues (Graves, Cha et al. 2014).

Although the disease is genetically mediated, it appears that non-genetic factors also influence the clinical manifestations (Graves, Rajakulendran et al. 2010, Gilbert, Graves et al. 2011).
Episodic ataxia type 1, ocular motor manifestations
Some of these patients exhibit down beat nystagmus (Jorge, Melancia et al. 2022).
Episodic ataxia type 1, inter-ictal symptoms
In between episodes, patients with EA1 often exhibit myokymia, but may also experience other symptoms, such as headache or pseudodystonia (Bhattacharjee, Deenadayalu et al. 2022).
Episodic ataxia type 1, treatments
Acetazolamide was fortuitously found to have some effect on symptoms of EA1 (Griggs, Moxley et al. 1978), and subsequently it was reported that approximately 50% of patients improve with acetazolamide (Baloh and Jen 2002).
There are a few reports of EA1 responding to phenytoin (Dressler and Benecke 2005).
Episodic ataxia type 2
Episodic ataxia type 2, epidemiology
The prevalence of EA2 has been estimated at 1 in 100,000 (Kotagal 2012). One report of several generations of a Korean family found decreasing age of onset with increasing number of trinucleotide repeats, suggesting anticipation (Choi, Yook et al. 2013).
Episodic ataxia type 2, clinical presentation
Episodes of EA2 usually begin in infancy or childhood. Some mutations appear to have much later onset; Cuenca-Leon and colleagues reported a patient in whom symptom onset was in the 6th decade (Cuenca-Leon, Banchs et al. 2009).
Episodes of EA2 can be triggered by stress, exercise and caffeine (Baloh and Jen 2002). The mechanism for these triggers is unclear, but animal models suggest that emotional and physiologic duress activate the locus coeruleus and trigger release of norepinephrine in the cerebellum, which has downstream effects of interfering with Purkinje cell function. This postulated mechanism is illustrated in the Figure below from Benarroch (Benarroch 2023).
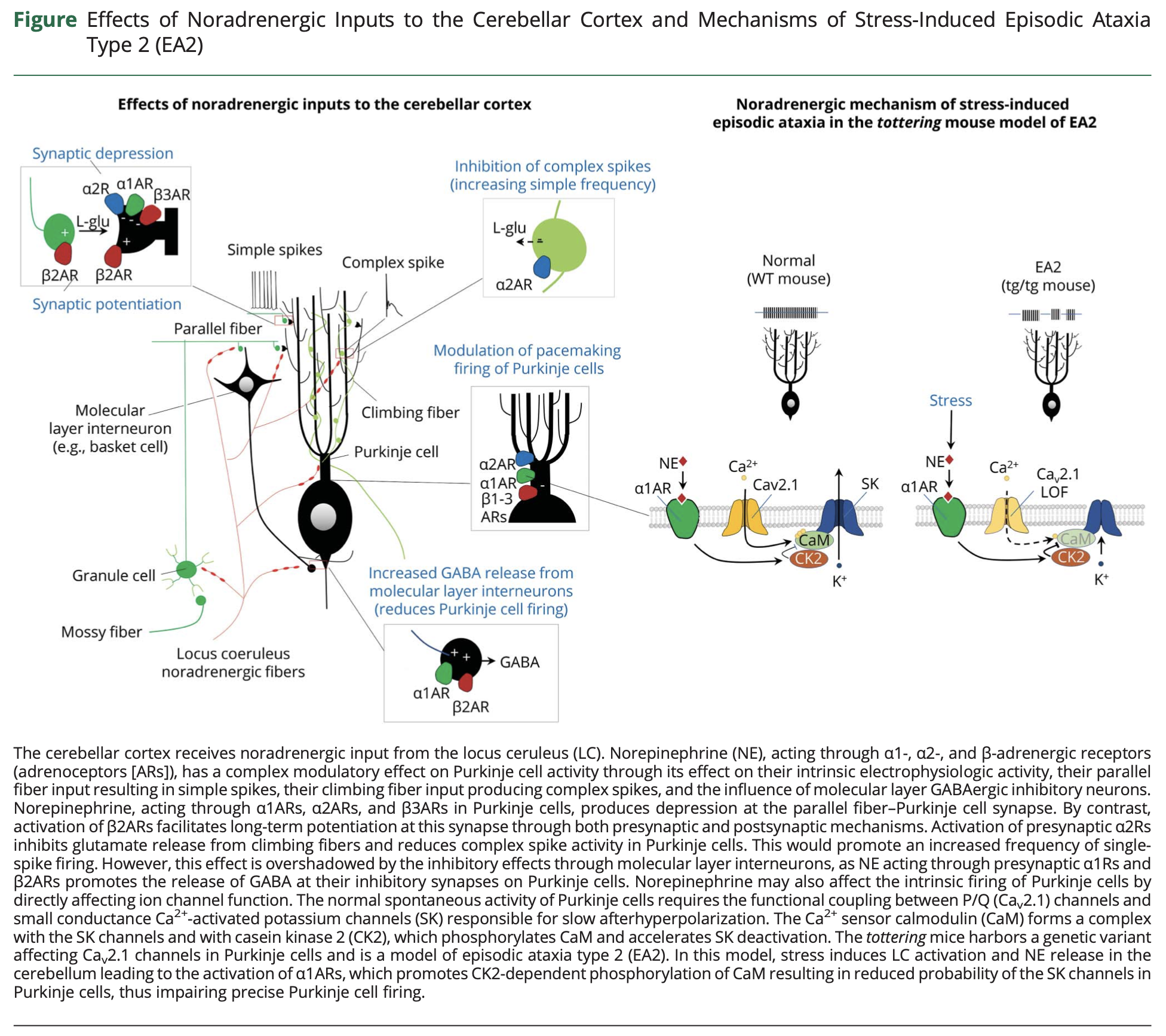
The frequency of the episodes in EA2 is extremely variable, ranging from a few per year to a few per week; they can last for hours (Kotagal 2012).
Ictal symptoms of EA2 include a sensation of vertigo, dysarthria and truncal ataxia (Kotagal 2012), and may be accompanied by headache (Dressler and Benecke 2005). Jen and colleagues (Jen, Kim et al. 2004) state that the most common neurologic symptoms are ataxia, vertigo and fluctuating weakness. Additional features are listed in the Table below.

Some patients describe experiencing vertigo without ataxia (Ling, Zhao et al. 2019).
The clinical presentation cannot be predicted from the specific mutation (discussed below) (Jen, Kim et al. 2004). Multiple patients in the same family with the same genetic mutation can have different clinical manifestations (Jung, Testard et al. 2010).
Episodic ataxia type 2, genetics
EA2 is inherited in an autosomal dominant fashion.
Linkage analysis identified a locus on chromosome 19p (Kramer, Yue et al. 1995, von Brederlow, Hahn et al. 1995) that was found to encode the CACNA1A calcium channel gene (Ophoff, Terwindt et al. 1996). Most of the CACNA1A mutations in EA2 are nonsense mutations leading to a truncated protein with loss of function (Kotagal 2012). However, over 30 mutations in this gene have been found to manifest as EA2, and these mutations are of various types, including nonsense mutations, missense mutations, deletions, an insertion and a donor splice site mutation (Jen, Kim et al. 2004).
A large number of mutations has been described in patients diagnosed with EA2 (Kramer, Yue et al. 1995, von Brederlow, Hahn et al. 1995, Ophoff, Terwindt et al. 1996, Denier, Ducros et al. 1999, Jen, Yue et al. 1999, Denier, Ducros et al. 2001, Guida, Trettel et al. 2001, Jen, Wan et al. 2001, Scoggan, Chandra et al. 2001, van den Maagdenberg, Kors et al. 2002, Hirose, Arayama et al. 2003, Subramony, Schott et al. 2003, Jen, Kim et al. 2004, Kaunisto, Harno et al. 2004, Spacey, Hildebrand et al. 2004, Eunson, Graves et al. 2005, Imbrici, Eunson et al. 2005, Spacey, Materek et al. 2005, Wan, Carr et al. 2005, Jeng, Chen et al. 2006, Kim, Kim et al. 2006, Graves, Imbrici et al. 2008, Jeng, Sun et al. 2008, Kaja, Van De Ven et al. 2008, Riant, Mourtada et al. 2008, Cuenca-Leon, Banchs et al. 2009, Labrum, Rajakulendran et al. 2009, Robbins, Lipton et al. 2009, Zafeiriou, Lehmann-Horn et al. 2009, Jung, Testard et al. 2010, Mantuano, Romano et al. 2010, Riant, Lescoat et al. 2010, Nikaido, Tachi et al. 2011, Wan, Mamsa et al. 2011, Hu, Jiang et al. 2013, Nachbauer, Nocker et al. 2014, Kinder, Ossig et al. 2015, Dahimene, Page et al. 2016, Maksemous, Roy et al. 2016, Pradotto, Mencarelli et al. 2016, Tomlinson, Tan et al. 2016, Dorgans, Salvi et al. 2017, Isaacs, Bradshaw et al. 2017, Petrovicova, Brozman et al. 2017, Sintas, Carreno et al. 2017, Sivak, Kurca et al. 2017, Balck, Tunc et al. 2018, Lance, Mossman et al. 2018, Maksemous, Smith et al. 2018, Ahuja, Rozen et al. 2019, Algahtani, Shirah et al. 2019, Nardello, Plicato et al. 2020, Verriello, Carrera et al. 2021, Verriello, Pauletto et al. 2021, Hommersom, van Prooije et al. 2022, Lipman, Fan et al. 2022, Nielsen, Asbjornsdottir et al. 2022, Xu, Wang et al. 2022).
The CACNA1A-encoded calcium channels are heavily expressed in Purkinje and granule cells in the cerebellum.
Other mutations in the CACNA1A gene manifest as other neurological disorders: familial hemiplegic migraine type 1, and spinocerebellar ataxia type 6 (Denier, Ducros et al. 1999, Jen 1999, Kotagal 2012, Pradotto, Mencarelli et al. 2016).
Episodic ataxia type 2, pathophysiology
The exact mechanism by which EA2 mutations provoke dysfunction of calcium channels is unclear. Possibilities include “reduced current density, increased rate of inactivation, and a shift in the voltage dependence of activation to more positive values” (Spacey, Hildebrand et al. 2004).
MR spectroscopy on patients with EA2 reported abnormal pH (alkalosis) in the cerebellum (Bain, O’Brien et al. 1992, Sappey-Marinier, Vighetto et al. 1999). Harno and colleagues (Harno, Heikkinen et al. 2005) speculate that acetazolamide may “stabilize the dysfunctioning P/Q‑type calcium channels, which express highly in the cerebellum.” Identification of abnormal metabolism in the cerebellar cortex seems logical given the clinical manifestations of EA2.
However, there is also some evidence of abnormalities in cerebral cortical function. Helmich and colleagues (Helmich, Siebner et al. 2010) studied six EA2 patients and 13 healthy controls with transcranial magnetic stimulation and reported that, “patients with episodic ataxia type 2 have an excessive increase in motor cortex excitability following a strong facilitatory input.” Indelicato and colleagues (Indelicato, Unterberger et al. 2021) studied 19 genetically confirmed EA2 patients with electroencephalography and found 12 (63%) were abnormal at baseline (outside of any attack), usually showing “lateralized intermittent slowly, mainly affecting the temporal region,” and 7 (37%) showed “interictal epileptic discharges.”
There is also some evidence of peripheral nerve dysfunction in EA2. Krishnan and colleagues (Krishnan, Bostock et al. 2008) reported on 3 members of a family with EA2 and found that, “Nerve excitability testing demonstrated significant abnormalities, with all patients outside the normal 95 % confidence limits in having a high rheobase and reduced early hyperpolarizing threshold electrotonus. On average there were also significant reductions in refractoriness, late sub excitability and early depolarizing threshold electrotonus.” Tomlinson and colleagues (Tomlinson, Tan et al. 2016) studied 8 patients from 2 families with EA2 and reported that, “All patients had similar excitability abnormalities, including a high electrical threshold and increased responses to hyperpolarizing and depolarizing currents in threshold electrotonus,” and 2 patients, showed “increased jitter in single-fibre EMG studies indicated unstable neuromuscular transmission.”
Episodic ataxia type 2, physical examination
If an EA2 patient is examined during an attack one may identify spontaneous nystagmus. Early in the disease the patient’s examination may be normal inter-ictally, but later there may be spontaneous down beat nystagmus even between attacks (Kotagal 2012) and gaze-evoked nystagmus (Bertholon, Chabrier et al. 2009).
Kim and colleagues (Kim, Kim et al. 2014) reported the case of an EA2 patient with rebound up beat nystagmus after lateral gaze holding — in effect, perverted rebound nystagmus.
Shervin Badv and Niksirat (Shervin Badv and Niksirat 2013) reported downward gaze palsy during attacks of a patient with EA2.
Episodic ataxia type 2, vestibular testing
Harno and colleagues (Harno, Hirvonen et al. 2004) studied three patients with genetically confirmed EA2 with nystagmography while the patients were off, and then back on, treatment with acetazolamide. The first patient while off treatment exhibited hypermetric saccades; this improved on treatment. The second patient, while off treatment, exhibited spontaneous left beat nystagmus of 5 deg/sec that increased with head turns; the nystagmus improved (but did not cease) on treatment. The third patient while off treatment exhibited spontaneous up beat nystagmus of 3 deg/sec and horizontal rebound nystagmus of 2 deg/sec.
The Figure below, from Harno and colleagues (Harno, Hirvonen et al. 2004), shows the spontaneous left beat nystagmus while off acetazolamide (panel A) and while on acetazolamide (panel B).
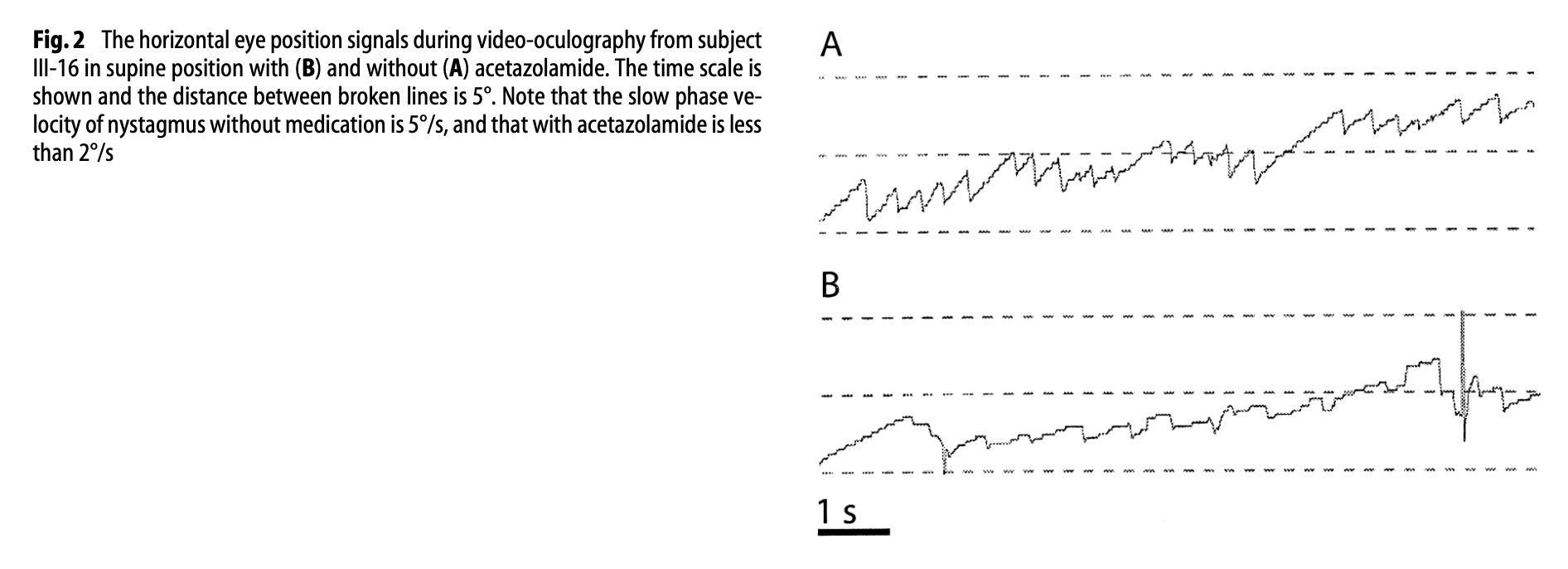
Figure : Nystagmographic tracings from a patient with genetically confirmed episodic ataxia type 2. Panel A shows spontaneous left beat nystagmus of 5 deg/sec while the patient was off treatment. Panel B shows reduction of the spontaneous left beat nystagmus to 2 deg/sec while the patient was on treatment with acetazolamide. From Harno et al. (2004).
Harno and colleagues (Harno, Hirvonen et al. 2004) also studied computerized dynamic posturography on the second patient, and reported that, “the sway velocities in posturography were pathologically increased without medication.”
Choi and colleagues (Harno, Hirvonen et al. 2004) studied 16 patients with EA2 inter-ictally and reported that more than half of the patients exhibited abnormalities in at least one of each vestibular test of video head impulse testing (vHIT), subjective visual vertical (SVV), ocular vestibular evoked myogenic potentials (oVEMP) or cervical vestibular evoked myogenic potentials (cVEMP). Specifically, “Patients with EA2 commonly showed abnormal VOR [vestibulo-ocular reflex] responses at least for one SCC [semicircular canal] with high-acceleration, high-frequency head impulses (14/16, 88%), and impaired visual-vestibular interaction (7/12, 58%). In response to low acceleration and frequency stimuli, the VOR gains were generally normal. The majority of EA2 patients had impairments in at least one of the otolith function tests (13/16, 81%): SVV [subjective visual vertical] tilt or variability (7/14, 50%), oVEMP [ocular vestibular evoked myogenic potentials] (8/15, 53%), and cVEMP [cervical vestibular evoked myogenic potentials] (4/16, 25%). Vestibular impairments are common in EA2 even during the interictal periods.” (Choi, Oh et al. 2022).
Another report of 4 patients observed, “deficits in tracking behavior with common features. Ocular tracking tended to result in hypermetric saccades at longer than normal latencies. Smooth pursuit tracking was absent in 1 patient and had lower than normal gain in the others” (Engel, Anderson et al. 2004).
Gordon and colleagues (Gordon, Caspi et al. 2008) studied four EA2 patients using scleral search coils. They reported that 2 (50%) exhibited “abnormal VOR” (vestibulo-ocular reflex) and “showed essentially no SP” (smooth pursuit).
Kipfer and colleagues (Kipfer, Jung et al. 2013) reported on a family of three EA2 patients who demonstrated slow horizontal saccades.
Kim and colleagues (Kim, Kim et al. 2022) reported the case of an EA2 patient with slow vertical saccades (both upward and downward).
Rucker and colleagues (Rucker, Jen et al. 2005) reported on two patients with EA2 who exhibited slowing of adducting saccades. Shimmura and colleagues (Shimmura, Uehara et al. 2017) described slow adducting movements during smooth pursuit in a patient with EA2.
Some investigators point out that there are some similarities in eye movement abnormalities between EA2 and spinocerebellar ataxia type 6 (Ying, Jen et al. 2001).
Episodic ataxia type 2, imaging
MR spectroscopy on patients with EA2 reported abnormal pH (alkalosis) in the cerebellum (Bain, O’Brien et al. 1992, Sappey-Marinier, Vighetto et al. 1999). Harno and colleagues (Harno, Heikkinen et al. 2005) speculate that acetazolamide may “stabilize the dysfunctioning P/Q‑type calcium channels, which express highly in the cerebellum.”
Episodic ataxia type 2, treatments
Acetazolamide remains the best studied treatment for EA2, with response rates ranging from 70% (Strupp, Zwergal et al. 2007) to 90% (Baloh and Jen 2002). This remains the best studied treatment.
One report described symptomatic improvement in EA2 through a combination of acetazolamide and valproate (Scoggan, Friedman et al. 2006).
There is also some literature regarding 4‑AP (4‑aminopyridine, also called dalfampridine) (Strupp, Kalla et al. 2004, Lohle, Schrempf et al. 2008, Strupp, Kalla et al. 2008, Alvina and Khodakhah 2010, Strupp, Kalla et al. 2011, Claassen, Teufel et al. 2013, Strupp, Teufel et al. 2017, Malamud and Otallah 2022). An extended release of 4‑AP called fampridine has also been studied (Muth, Teufel et al. 2021).
One report advocated the combination of 4‑AP plus topiramate in patients unresponsive to acetazolamide (Gonzalez-Mingot, Lopez-Ortega et al. 2022).
Other proposed treatments have included chlorzoxazone (Alvina and Khodakhah 2010), levetiracetam (Lee, Jang et al. 2017, Na and Kim 2021) and flunarizine (Yuan, Zheng et al. 2022).
Episodic ataxia type 3
In 2001 Steckley and colleagues (Steckley, Ebers et al. 2001) reported on a large Canadian family of Mennonite ancestry. In this kindred there was a broad age range of symptom onset (1 – 42 years). Episodes were sometimes provoked by movement (kinesiogenic), involved symptoms of vertigo, nausea and tinnitus, and tended to last minutes, rarely hours, and responded to acetazolamide (Steckley, Ebers et al. 2001). Linkage analysis identified a locus on chromosome 1q42 (Cader, Steckley et al. 2005), but the specific gene has not yet been identified.
Episodic ataxia type 4
Episodic ataxia type 4, also called periodic vestibulocerebellar ataxia, is another autosomal dominant ataxia originally described in two families from North Carolina (Damji, Allingham et al. 1996). Its symptoms are similar to those of EA2 (Kotagal 2012), though the age of onset is later (3rd – 6th decade) and the attacks do not respond to acetazolamide.
Episodic ataxia type 5
Episodic ataxia type 5 was described by Escayg and colleagues (Escayg, De Waard et al. 2000) who reported on a French Canadian kindred whose members suffered from attacks very similar to EA2, although the age of onset was somewhat later (3rd – 4th decade); symptoms included vertigo, ataxia and dysarthria, lasting hours at a time, improved with acetazolamide (Kotagal 2012). The disorder is linked to a mutation in a gene for a different calcium channel (CACNB4).
Episodic ataxia type 6
Episodic ataxia type 6 was described in a single child in a report by Jen and colleagues (Jen, Wan et al. 2005). Aside from symptoms of ataxia, the episodes are different in that they involve hemiplegia and seizures. The disease has been attributed to a mutation in an astrocyte glutamate transporter (Kotagal 2012).
References
Ahuja AS, Rozen TD, Atwal PS (2019) A sleep modulated Channelopathy: a novel CACNA1A pathogenic variant identified in episodic Ataxia type 2 and a potential link to sleep alleviated migraine. BMC Neurol 19: 246. doi: 10.1186/s12883-019-1491-3
Algahtani H, Shirah B, Algahtani R, Al-Qahtani MH, Abdulkareem AA, Naseer MI (2019) A novel mutation in CACNA1A gene in a Saudi female with episodic ataxia type 2 with no response to acetazolamide or 4-aminopyridine. Intractable Rare Dis Res 8: 67-71. doi: 10.5582/irdr.2018.01133
Alvina K, Khodakhah K (2010a) KCa channels as therapeutic targets in episodic ataxia type-2. J Neurosci 30: 7249-57. doi: 10.1523/JNEUROSCI.6341-09.2010
Alvina K, Khodakhah K (2010b) The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J Neurosci 30: 7258-68. doi: 10.1523/JNEUROSCI.3582-09.2010
Bain PG, O’Brien MD, Keevil SF, Porter DA (1992) Familial periodic cerebellar ataxia: a problem of cerebellar intracellular pH homeostasis. Ann Neurol 31: 147-54. doi: 10.1002/ana.410310205
Balck A, Tunc S, Schmitz J, Hollstein R, Kaiser FJ, Bruggemann N (2018) A Novel Frameshift CACNA1A Mutation Causing Episodic Ataxia Type 2. Cerebellum 17: 504-506. doi: 10.1007/s12311-018-0931-8
Baloh RW, Jen JC (2002) Genetics of familial episodic vertigo and ataxia. Ann N Y Acad Sci 956: 338-45.
Benarroch E (2023) What Is the Role of Norepinephrine in Cerebellar Modulation and Stress-Induced Episodic Ataxia? Neurology 100: 383-386. doi: 10.1212/WNL.0000000000206882
Bertholon P, Chabrier S, Riant F, Tournier-Lasserve E, Peyron R (2009) Episodic ataxia type 2: unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J Neurol Neurosurg Psychiatry 80: 1289-92. doi: 10.1136/jnnp.2008.159103
Bhattacharjee S, Deenadayalu A, Paramanandam V (2022) Interictal Headache, Pseudodystonia, and Persistent Ataxia in Episodic Ataxia Type 1 Due to a Novel KCNA1 Gene Mutation. Mov Disord Clin Pract 9: 272-274. doi: 10.1002/mdc3.13381
Bretschneider F, Wrisch A, Lehmann-Horn F, Grissmer S (1999) Expression in mammalian cells and electrophysiological characterization of two mutant Kv1.1 channels causing episodic ataxia type 1 (EA-1). Eur J Neurosci 11: 2403-12. doi: 10.1046/j.1460-9568.1999.00659.x
Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M (1994) Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8: 136-40. doi: 10.1038/ng1094-136
Cader MZ, Steckley JL, Dyment DA, McLachlan RS, Ebers GC (2005) A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia. Neurology 65: 156-8. doi: 10.1212/01.wnl.0000167186.05465.7c
Chen SH, Fu SJ, Huang JJ, Tang CY (2016) The episodic ataxia type 1 mutation I262T alters voltage-dependent gating and disrupts protein biosynthesis of human Kv1.1 potassium channels. Sci Rep 6: 19378. doi: 10.1038/srep19378
Choi JH, Oh EH, Choi SY, Kim HJ, Lee SK, Choi JY, Kim JS, Choi KD (2022) Vestibular impairments in episodic ataxia type 2. J Neurol 269: 2687-2695. doi: 10.1007/s00415-021-10856-4
Choi KD, Yook JW, Kim MJ, Kim HS, Park YE, Kim JS, Choi JH, Shin JH, Kim DS (2013) Possible anticipation associated with a novel splice site mutation in episodic ataxia type 2. Neurol Sci 34: 1629-32. doi: 10.1007/s10072-013-1298-8
Claassen J, Teufel J, Kalla R, Spiegel R, Strupp M (2013) Effects of dalfampridine on attacks in patients with episodic ataxia type 2: an observational study. J Neurol 260: 668-9. doi: 10.1007/s00415-012-6764-3
Cuenca-Leon E, Banchs I, Serra SA, Latorre P, Fernandez-Castillo N, Corominas R, Valverde MA, Volpini V, Fernandez-Fernandez JM, Macaya A, Cormand B (2009) Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J Neurol Sci 280: 10-4. doi: 10.1016/j.jns.2009.01.005
D’Adamo MC, Gallenmuller C, Servettini I, Hartl E, Tucker SJ, Arning L, Biskup S, Grottesi A, Guglielmi L, Imbrici P, Bernasconi P, Di Giovanni G, Franciolini F, Catacuzzeno L, Pessia M, Klopstock T (2014) Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front Physiol 5: 525. doi: 10.3389/fphys.2014.00525
D’Adamo MC, Imbrici P, Sponcichetti F, Pessia M (1999) Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J 13: 1335-45. doi: 10.1096/fasebj.13.11.1335
D’Adamo MC, Liu Z, Adelman JP, Maylie J, Pessia M (1998) Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J 17: 1200-7. doi: 10.1093/emboj/17.5.1200
Dahimene S, Page KM, Nieto-Rostro M, Pratt WS, D’Arco M, Dolphin AC (2016) A CaV2.1 N-terminal fragment relieves the dominant-negative inhibition by an Episodic ataxia 2 mutant. Neurobiol Dis 93: 243-56. doi: 10.1016/j.nbd.2016.05.020
Damji KF, Allingham RR, Pollock SC, Small K, Lewis KE, Stajich JM, Yamaoka LH, Vance JM, Pericak-Vance MA (1996) Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol 53: 338-44. doi: 10.1001/archneur.1996.00550040074016
Denier C, Ducros A, Durr A, Eymard B, Chassande B, Tournier-Lasserve E (2001) Missense CACNA1A mutation causing episodic ataxia type 2. Arch Neurol 58: 292-5. doi: 10.1001/archneur.58.2.292
Denier C, Ducros A, Vahedi K, Joutel A, Thierry P, Ritz A, Castelnovo G, Deonna T, Gerard P, Devoize JL, Gayou A, Perrouty B, Soisson T, Autret A, Warter JM, Vighetto A, Van Bogaert P, Alamowitch S, Roullet E, Tournier-Lasserve E (1999) High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 52: 1816-21. doi: 10.1212/wnl.52.9.1816
Dorgans K, Salvi J, Bertaso F, Bernard L, Lory P, Doussau F, Mezghrani A (2017) Characterization of the dominant inheritance mechanism of Episodic Ataxia type 2. Neurobiol Dis 106: 110-123. doi: 10.1016/j.nbd.2017.07.004
Dressler D, Benecke R (2005) Diagnosis and management of acute movement disorders. J Neurol 252: 1299-306. doi: 10.1007/s00415-005-0006-x
Engel KC, Anderson JH, Gomez CM, Soechting JF (2004) Deficits in ocular and manual tracking due to episodic ataxia type 2. Mov Disord 19: 778-787. doi: 10.1002/mds.20121
Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, Johnston J, Baloh R, Sander T, Meisler MH (2000) Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet 66: 1531-9. doi: 10.1086/302909
Eunson LH, Graves TD, Hanna MG (2005) New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology 65: 308-10. doi: 10.1212/01.wnl.0000169020.82223.dd
Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, Avoni P, McWilliam RC, Stephenson JB, Hanna MG, Kullmann DM, Spauschus A (2000) Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 48: 647-56.
Ferrick-Kiddie EA, Rosenthal JJ, Ayers GD, Emeson RB (2017) Mutations underlying Episodic Ataxia type-1 antagonize Kv1.1 RNA editing. Sci Rep 7: 41095. doi: 10.1038/srep41095
Gilbert GJ, Graves TD, Kullmann DM (2011) Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 76: 490; author reply 490. doi: 10.1212/WNL.0b013e3182068ea0
Gonzalez-Mingot C, Lopez-Ortega R, Brieva-Ruiz L (2022) The efficacy of combining topiramate and 4-aminopyridine to reduce relapses and interictal progression in two cases of episodic ataxia type 2. Neurol Sci 43: 5099-5101. doi: 10.1007/s10072-022-06144-2
Gordon CR, Caspi A, Levite R, Zivotofsky AZ (2008) Mechanisms of vestibulo-ocular reflex (VOR) cancellation in spinocerebellar ataxia type 3 (SCA-3) and episodic ataxia type 2 (EA-2). Prog Brain Res 171: 519-25. doi: 10.1016/S0079-6123(08)00674-2
Graves TD, Cha YH, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, Bundy BN, Jen JC, Baloh RW, Hanna MG, Investigators C (2014) Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain 137: 1009-18. doi: 10.1093/brain/awu012
Graves TD, Imbrici P, Kors EE, Terwindt GM, Eunson LH, Frants RR, Haan J, Ferrari MD, Goadsby PJ, Hanna MG, van den Maagdenberg AM, Kullmann DM (2008) Premature stop codons in a facilitating EF-hand splice variant of CaV2.1 cause episodic ataxia type 2. Neurobiol Dis 32: 10-5. doi: 10.1016/j.nbd.2008.06.002
Graves TD, Rajakulendran S, Zuberi SM, Morris HR, Schorge S, Hanna MG, Kullmann DM (2010) Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 75: 367-72. doi: 10.1212/WNL.0b013e3181ea9ee3
Griggs RC, Moxley RT, 3rd, Lafrance RA, McQuillen J (1978) Hereditary paroxysmal ataxia: response to acetazolamide. Neurology 28: 1259-64. doi: 10.1212/wnl.28.12.1259
Guida S, Trettel F, Pagnutti S, Mantuano E, Tottene A, Veneziano L, Fellin T, Spadaro M, Stauderman K, Williams M, Volsen S, Ophoff R, Frants R, Jodice C, Frontali M, Pietrobon D (2001) Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am J Hum Genet 68: 759-64. doi: 10.1086/318804
Hanson PA, Martinez LB, Cassidy R (1977) Contractures, continuous muscle discharges, and titubation. Ann Neurol 1: 120-4. doi: 10.1002/ana.410010203
Harno H, Heikkinen S, Kaunisto MA, Kallela M, Hakkinen AM, Wessman M, Farkkila M, Lundbom N (2005) Decreased cerebellar total creatine in episodic ataxia type 2: a 1H MRS study. Neurology 64: 542-4. doi: 10.1212/01.WNL.0000150589.26350.3D
Harno H, Hirvonen T, Kaunisto MA, Aalto H, Levo H, Isotalo E, Somer H, Kallela M, Palotie A, Wessman M, Farkkila M (2004) Acetazolamide improves neurotological abnormalities in a family with episodic ataxia type 2 (EA-2). J Neurol 251: 232-4. doi: 10.1007/s00415-004-0299-1
Heeroma JH, Henneberger C, Rajakulendran S, Hanna MG, Schorge S, Kullmann DM (2009) Episodic ataxia type 1 mutations differentially affect neuronal excitability and transmitter release. Dis Model Mech 2: 612-9. doi: 10.1242/dmm.003582
Helmich RC, Siebner HR, Giffin N, Bestmann S, Rothwell JC, Bloem BR (2010) The dynamic regulation of cortical excitability is altered in episodic ataxia type 2. Brain 133: 3519-29. doi: 10.1093/brain/awq315
Hirose H, Arayama T, Takita J, Igarashi T, Hayashi Y, Nagao Y (2003) A family of episodic ataxia type 2: no evidence of genetic linkage to the CACNA1A gene. Int J Mol Med 11: 187-9.
Hommersom MP, van Prooije TH, Pennings M, Schouten MI, van Bokhoven H, Kamsteeg EJ, van de Warrenburg BPC (2022) The complexities of CACNA1A in clinical neurogenetics. J Neurol 269: 3094-3108. doi: 10.1007/s00415-021-10897-9
Hu Y, Jiang H, Wang Q, Xie Z, Pan S (2013) Identification of a novel nonsense mutation p.Tyr1957Ter of CACNA1A in a Chinese family with episodic ataxia 2. PLoS One 8: e56362. doi: 10.1371/journal.pone.0056362
Imbrici P, D’Adamo MC, Grottesi A, Biscarini A, Pessia M (2011) Episodic ataxia type 1 mutations affect fast inactivation of K+ channels by a reduction in either subunit surface expression or affinity for inactivation domain. Am J Physiol Cell Physiol 300: C1314-22. doi: 10.1152/ajpcell.00456.2010
Imbrici P, D’Adamo MC, Kullmann DM, Pessia M (2006) Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur J Neurosci 24: 3073-83. doi: 10.1111/j.1460-9568.2006.05186.x
Imbrici P, Eunson LH, Graves TD, Bhatia KP, Wadia NH, Kullmann DM, Hanna MG (2005) Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology 65: 944-6. doi: 10.1212/01.wnl.0000176069.64200.28
Imbrici P, Gualandi F, D’Adamo MC, Masieri MT, Cudia P, De Grandis D, Mannucci R, Nicoletti I, Tucker SJ, Ferlini A, Pessia M (2008) A novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1. Neuroscience 157: 577-87. doi: 10.1016/j.neuroscience.2008.09.022
Indelicato E, Unterberger I, Nachbauer W, Eigentler A, Amprosi M, Zeiner F, Haberlandt E, Kaml M, Gizewski E, Boesch S (2021) The electrophysiological footprint of CACNA1A disorders. J Neurol 268: 2493-2505. doi: 10.1007/s00415-021-10415-x
Isaacs DA, Bradshaw MJ, Brown K, Hedera P (2017) Case report of novel CACNA1A gene mutation causing episodic ataxia type 2. SAGE Open Med Case Rep 5: 2050313X17706044. doi: 10.1177/2050313X17706044
Jen J (1999) Calcium channelopathies in the central nervous system. Curr Opin Neurobiol 9: 274-80. doi: 10.1016/s0959-4388(99)80040-3
Jen J, Kim GW, Baloh RW (2004) Clinical spectrum of episodic ataxia type 2. Neurology 62: 17-22. doi: 10.1212/01.wnl.0000101675.61074.50
Jen J, Wan J, Graves M, Yu H, Mock AF, Coulin CJ, Kim G, Yue Q, Papazian DM, Baloh RW (2001) Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology 57: 1843-8. doi: 10.1212/wnl.57.10.1843
Jen J, Yue Q, Nelson SF, Yu H, Litt M, Nutt J, Baloh RW (1999) A novel nonsense mutation in CACNA1A causes episodic ataxia and hemiplegia. Neurology 53: 34-7. doi: 10.1212/wnl.53.1.34
Jen JC, Wan J, Palos TP, Howard BD, Baloh RW (2005) Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65: 529-34. doi: 10.1212/01.wnl.0000172638.58172.5a
Jeng CJ, Chen YT, Chen YW, Tang CY (2006) Dominant-negative effects of human P/Q-type Ca2+ channel mutations associated with episodic ataxia type 2. Am J Physiol Cell Physiol 290: C1209-20. doi: 10.1152/ajpcell.00247.2005
Jeng CJ, Sun MC, Chen YW, Tang CY (2008) Dominant-negative effects of episodic ataxia type 2 mutations involve disruption of membrane trafficking of human P/Q-type Ca2+ channels. J Cell Physiol 214: 422-33. doi: 10.1002/jcp.21216
Jorge A, Melancia D, Figueiredo C, Galego O, Oliveira J, Martins AI, Lemos J (2022) Downbeat Nystagmus in Episodic Ataxia Type 1 Associated with a Novel KCNA1 Mutation. Mov Disord 37: 430-432. doi: 10.1002/mds.28843
Jung J, Testard H, Tournier-Lasserve E, Riant F, Vallet AE, Berroir S, Broussolle E (2010) Phenotypic variability of episodic ataxia type 2 mutations: a family study. Eur Neurol 64: 114-6. doi: 10.1159/000315145
Kaja S, Van De Ven RC, Frants RR, Ferrari MD, Van Den Maagdenberg AM, Plomp JJ (2008) Reduced ACh release at neuromuscular synapses of heterozygous leaner Ca(v)2.1-mutant mice. Synapse 62: 337-44. doi: 10.1002/syn.20490
Karalok ZS, Megaro A, Cenciarini M, Guven A, Hasan SM, Taskin BD, Imbrici P, Ceylaner S, Pessia M, D’Adamo MC (2018) Identification of a New de Novo Mutation Underlying Regressive Episodic Ataxia Type I. Front Neurol 9: 587. doi: 10.3389/fneur.2018.00587
Kaunisto MA, Harno H, Kallela M, Somer H, Sallinen R, Hamalainen E, Miettinen PJ, Vesa J, Orpana A, Palotie A, Farkkila M, Wessman M (2004) Novel splice site CACNA1A mutation causing episodic ataxia type 2. Neurogenetics 5: 69-73. doi: 10.1007/s10048-003-0161-0
Kim HJ, Kim JS, Choi JH, Shin JH, Choi KD, Zee DS (2014) Rebound upbeat nystagmus after lateral gaze in episodic ataxia type 2. Cerebellum 13: 411-3. doi: 10.1007/s12311-014-0547-6
Kim JM, Kim JS, Ki CS, Jeon BS (2006) Episodic Ataxia Type 2 due to a Deletion Mutation in the CACNA1A Gene in a Korean Family. J Clin Neurol 2: 268-71. doi: 10.3988/jcn.2006.2.4.268
Kim S, Kim S, Lee S, Kim HJ (2022) Vertical Saccadic Slowing in Episodic Ataxia Type 2. J Clin Neurol 18: 726-728. doi: 10.3988/jcn.2022.18.6.726
Kinder S, Ossig C, Wienecke M, Beyer A, von der Hagen M, Storch A, Smitka M (2015) Novel frameshift mutation in the CACNA1A gene causing a mixed phenotype of episodic ataxia and familiar hemiplegic migraine. Eur J Paediatr Neurol 19: 72-4. doi: 10.1016/j.ejpn.2014.10.005
Kipfer S, Jung S, Lemke JR, Kipfer-Kauer A, Howell JP, Kaelin-Lang A, Nyffeler T, Gutbrod K, Abicht A, Muri RM (2013) Novel CACNA1A mutation(s) associated with slow saccade velocities. J Neurol 260: 3010-4. doi: 10.1007/s00415-013-7099-4
Klaas JP, Burkholder DB, Singer W, Boes CJ (2013) Harry Lee Parker and paroxysmal dysarthria and ataxia. Neurology 80: 311-4. doi: 10.1212/WNL.0b013e31827dec0f
Klein A, Boltshauser E, Jen J, Baloh RW (2004) Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics 35: 147-9. doi: 10.1055/s-2004-817921
Knight MA, Storey E, McKinlay Gardner RJ, Hand P, Forrest SM (2000) Identification of a novel missense mutation L329I in the episodic ataxia type 1 gene KCNA1–a challenging problem. Hum Mutat 16: 374. doi: 10.1002/1098-1004(200010)16:4<374::AID-HUMU15>3.0.CO;2-4
Kotagal V (2012) Acetazolamide-responsive ataxia. Semin Neurol 32: 533-7. doi: 10.1055/s-0033-1334475
Kramer PL, Yue Q, Gancher ST, Nutt JG, Baloh R, Smith E, Browne D, Bussey K, Lovrien E, Nelson S, et al. (1995) A locus for the nystagmus-associated form of episodic ataxia maps to an 11-cM region on chromosome 19p. Am J Hum Genet 57: 182-5.
Krishnan AV, Bostock H, Ip J, Hayes M, Watson S, Kiernan MC (2008) Axonal function in a family with episodic ataxia type 2 due to a novel mutation. J Neurol 255: 750-5. doi: 10.1007/s00415-008-0794-x
Labrum RW, Rajakulendran S, Graves TD, Eunson LH, Bevan R, Sweeney MG, Hammans SR, Tubridy N, Britton T, Carr LJ, Ostergaard JR, Kennedy CR, Al-Memar A, Kullmann DM, Schorge S, Temple K, Davis MB, Hanna MG (2009) Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: implications for diagnostic testing. J Med Genet 46: 786-91. doi: 10.1136/jmg.2009.067967
Lance S, Mossman S, Poke G (2018) A Novel CACNA1A Nonsense Variant [c.4054C>T (p.Arg1352(⁎))] Causing Episodic Ataxia Type 2. Case Rep Neurol Med 2018: 5802650. doi: 10.1155/2018/5802650
Lassche S, Lainez S, Bloem BR, van de Warrenburg BP, Hofmeijer J, Lemmink HH, Hoenderop JG, Bindels RJ, Drost G (2014) A novel KCNA1 mutation causing episodic ataxia type I. Muscle Nerve 50: 289-91. doi: 10.1002/mus.24242
Lee GB, Kim GY, Jeong IH, Kim N, Kim JW (2021) A Novel KCNA1 Mutation in an Episodic Ataxia Type 1 Patient with Asterixis and Falls. J Clin Neurol 17: 333-335. doi: 10.3988/jcn.2021.17.2.333
Lee H, Jang DH, Jang JH, Kim T (2017) Effectiveness of levetiracetam in an acetazolamide-unresponsive patient with episodic ataxia type 2 by a novel CACNA1A nonsense mutation. Eur J Neurol 24: e43-e44. doi: 10.1111/ene.13327
Ling X, Zhao DH, Zhao J, Shen B, Yang X (2019) Episodic ataxia type 2 characterised by recurrent dizziness/vertigo: a report of four cases. Int J Neurosci 129: 103-109. doi: 10.1080/00207454.2018.1486829
Lipman AR, Fan X, Shen Y, Chung WK (2022) Clinical and genetic characterization of CACNA1A-related disease. Clin Genet 102: 288-295. doi: 10.1111/cge.14180
Litt M, Kramer P, Browne D, Gancher S, Brunt ER, Root D, Phromchotikul T, Dubay CJ, Nutt J (1994) A gene for episodic ataxia/myokymia maps to chromosome 12p13. Am J Hum Genet 55: 702-9.
Lohle M, Schrempf W, Wolz M, Reichmann H, Storch A (2008) Potassium channel blocker 4-aminopyridine is effective in interictal cerebellar symptoms in episodic ataxia type 2–a video case report. Mov Disord 23: 1314-6. doi: 10.1002/mds.22071
Maksemous N, Roy B, Smith RA, Griffiths LR (2016) Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol Genet Genomic Med 4: 211-22. doi: 10.1002/mgg3.196
Maksemous N, Smith RA, Sutherland HG, Sampaio H, Griffiths LR (2018) Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. Int J Mol Sci 19. doi: 10.3390/ijms19103113
Malamud E, Otallah SI (2022) Use of Dalfampridine in a Young Child with Episodic Ataxia Type 2. Child Neurol Open 9: 2329048X221075447. doi: 10.1177/2329048X221075447
Manganas LN, Akhtar S, Antonucci DE, Campomanes CR, Dolly JO, Trimmer JS (2001) Episodic ataxia type-1 mutations in the Kv1.1 potassium channel display distinct folding and intracellular trafficking properties. J Biol Chem 276: 49427-34. doi: 10.1074/jbc.M109325200
Mantuano E, Romano S, Veneziano L, Gellera C, Castellotti B, Caimi S, Testa D, Estienne M, Zorzi G, Bugiani M, Rajabally YA, Barcina MJ, Servidei S, Panico A, Frontali M, Mariotti C (2010) Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci 291: 30-6. doi: 10.1016/j.jns.2010.01.010
Maylie B, Bissonnette E, Virk M, Adelman JP, Maylie JG (2002) Episodic ataxia type 1 mutations in the human Kv1.1 potassium channel alter hKvbeta 1-induced N-type inactivation. J Neurosci 22: 4786-93. doi: 10.1523/JNEUROSCI.22-12-04786.2002
Mestre TA, Manole A, MacDonald H, Riazi S, Kraeva N, Hanna MG, Lang AE, Mannikko R, Yoon G (2016) A novel KCNA1 mutation in a family with episodic ataxia and malignant hyperthermia. Neurogenetics 17: 245-249. doi: 10.1007/s10048-016-0486-0
Muth C, Teufel J, Schols L, Synofzik M, Franke C, Timmann D, Mansmann U, Strupp M (2021) Fampridine and Acetazolamide in EA2 and Related Familial EA: A Prospective Randomized Placebo-Controlled Trial. Neurol Clin Pract 11: e438-e446. doi: 10.1212/CPJ.0000000000001017
Na S, Kim T (2021) Efficacy of levetiracetam in patients with episodic ataxia type 2 caused by CACNA1A mutation: three case reports. Neurol Sci 42: 3897-3899. doi: 10.1007/s10072-021-05368-y
Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, Eigentler A, Schneider R, Poewe W, Delazer M, Boesch S (2014) Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature. J Neurol 261: 983-91. doi: 10.1007/s00415-014-7310-2
Nardello R, Plicato G, Mangano GD, Gennaro E, Mangano S, Brighina F, Raieli V, Fontana A (2020) Two distinct phenotypes, hemiplegic migraine and episodic Ataxia type 2, caused by a novel common CACNA1A variant. BMC Neurol 20: 155. doi: 10.1186/s12883-020-01704-5
Nielsen EN, Asbjornsdottir B, Moller LB, Nielsen JE, Lindquist SG (2022) Episodic ataxia type 2 (EA2) with interictal myokymia and focal dystonia. Cold Spring Harb Mol Case Stud 8. doi: 10.1101/mcs.a006236
Nikaido K, Tachi N, Ohya K, Wada T, Tsutsumi H (2011) New mutation of CACNA1A gene in episodic ataxia type 2. Pediatr Int 53: 415-6. doi: 10.1111/j.1442-200X.2011.03390.x
Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87: 543-52. doi: 10.1016/s0092-8674(00)81373-2
Parker HL (1946) Periodic ataxia. Collect Papers Mayo Clinic Mayo Found 38: 642-5.
Peters CJ, Werry D, Gill HS, Accili EA, Fedida D (2011) Mechanism of accelerated current decay caused by an episodic ataxia type-1-associated mutant in a potassium channel pore. J Neurosci 31: 17449-59. doi: 10.1523/JNEUROSCI.2940-11.2011
Petitjean D, Kalstrup T, Zhao J, Blunck R (2015) A Disease Mutation Causing Episodic Ataxia Type I in the S1 Links Directly to the Voltage Sensor and the Selectivity Filter in Kv Channels. J Neurosci 35: 12198-206. doi: 10.1523/JNEUROSCI.1419-15.2015
Petrovicova A, Brozman M, Kurca E, Gobo T, Dluha J, Kalmarova K, Nosal V, Hikkelova M, Krajciova A, Burjanivova T, Sivak S (2017) Novel missense variant of CACNA1A gene in a Slovak family with episodic ataxia type 2. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 161: 107-110. doi: 10.5507/bp.2016.066
Pradotto L, Mencarelli M, Bigoni M, Milesi A, Di Blasio A, Mauro A (2016) Episodic ataxia and SCA6 within the same family due to the D302N CACNA1A gene mutation. J Neurol Sci 371: 81-84. doi: 10.1016/j.jns.2016.10.029
Rajakulendran S, Schorge S, Kullmann DM, Hanna MG (2007) Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics 4: 258-66. doi: 10.1016/j.nurt.2007.01.010
Riant F, Lescoat C, Vahedi K, Kaphan E, Toutain A, Soisson T, Wiener-Vacher SR, Tournier-Lasserve E (2010) Identification of CACNA1A large deletions in four patients with episodic ataxia. Neurogenetics 11: 101-6. doi: 10.1007/s10048-009-0208-y
Riant F, Mourtada R, Saugier-Veber P, Tournier-Lasserve E (2008) Large CACNA1A deletion in a family with episodic ataxia type 2. Arch Neurol 65: 817-20. doi: 10.1001/archneur.65.6.817
Robbins MS, Lipton RB, Laureta EC, Grosberg BM (2009) CACNA1A nonsense mutation is associated with basilar-type migraine and episodic ataxia type 2. Headache 49: 1042-6. doi: 10.1111/j.1526-4610.2009.01464.x
Rucker JC, Jen J, Stahl JS, Natesan N, Baloh RW, Leigh RJ (2005) Internuclear ophthalmoparesis in episodic ataxia type 2. Ann N Y Acad Sci 1039: 571-4. doi: 10.1196/annals.1325.070
Sappey-Marinier D, Vighetto A, Peyron R, Broussolle E, Bonmartin A (1999) Phosphorus and proton magnetic resonance spectroscopy in episodic ataxia type 2. Ann Neurol 46: 256-9. doi: 10.1002/1531-8249(199908)46:2<256::aid-ana17>3.0.co;2-6
Scoggan KA, Chandra T, Nelson R, Hahn AF, Bulman DE (2001) Identification of two novel mutations in the CACNA1A gene responsible for episodic ataxia type 2. J Med Genet 38: 249-53. doi: 10.1136/jmg.38.4.249
Scoggan KA, Friedman JH, Bulman DE (2006) CACNA1A mutation in a EA-2 patient responsive to acetazolamide and valproic acid. Can J Neurol Sci 33: 68-72. doi: 10.1017/s0317167100004728
Shervin Badv R, Niksirat A (2013) Downward vertical gaze palsy as a prominent manifestation of episodic ataxia type 2: a case report. Iran J Child Neurol 7: 58-60.
Shimmura M, Uehara T, Yamashita K, Shigeto H, Yamasaki R, Ishikawa K, Kira JI (2017) Slowed abduction during smooth pursuit eye movement in episodic ataxia type 2 with a novel CACNA1A mutation. J Neurol Sci 381: 4-6. doi: 10.1016/j.jns.2017.07.040
Shook SJ, Mamsa H, Jen JC, Baloh RW, Zhou L (2008) Novel mutation in KCNA1 causes episodic ataxia with paroxysmal dyspnea. Muscle Nerve 37: 399-402. doi: 10.1002/mus.20904
Sintas C, Carreno O, Fernandez-Castillo N, Corominas R, Vila-Pueyo M, Toma C, Cuenca-Leon E, Barroeta I, Roig C, Volpini V, Macaya A, Cormand B (2017) Mutation Spectrum in the CACNA1A Gene in 49 Patients with Episodic Ataxia. Sci Rep 7: 2514. doi: 10.1038/s41598-017-02554-x
Sivak S, Kurca E, Krajciova A, Hikkelova M, Simko J, Misovicova N, Kantorova E, Turcanova-Koprusakova M, Burjanivova T, Cierny D, Nosal V (2017) Novel missense variant of CACNA1A gene: A case report of a family with episodic ataxia type 2. J Neurol Sci 376: 119-120. doi: 10.1016/j.jns.2017.03.008
Spacey SD, Hildebrand ME, Materek LA, Bird TD, Snutch TP (2004) Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann Neurol 56: 213-20. doi: 10.1002/ana.20169
Spacey SD, Materek LA, Szczygielski BI, Bird TD (2005) Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol 62: 314-6. doi: 10.1001/archneur.62.2.314
Steckley JL, Ebers GC, Cader MZ, McLachlan RS (2001) An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology 57: 1499-502. doi: 10.1212/wnl.57.8.1499
Strupp M, Kalla R, Claassen J, Adrion C, Mansmann U, Klopstock T, Freilinger T, Neugebauer H, Spiegel R, Dichgans M, Lehmann-Horn F, Jurkat-Rott K, Brandt T, Jen JC, Jahn K (2011) A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 77: 269-75. doi: 10.1212/WNL.0b013e318225ab07
Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T (2004) Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology 62: 1623-5. doi: 10.1212/01.wnl.0000125691.74109.53
Strupp M, Kalla R, Glasauer S, Wagner J, Hufner K, Jahn K, Brandt T (2008) Aminopyridines for the treatment of cerebellar and ocular motor disorders. Prog Brain Res 171: 535-41. doi: 10.1016/S0079-6123(08)00676-6
Strupp M, Teufel J, Zwergal A, Schniepp R, Khodakhah K, Feil K (2017) Aminopyridines for the treatment of neurologic disorders. Neurol Clin Pract 7: 65-76. doi: 10.1212/CPJ.0000000000000321
Strupp M, Zwergal A, Brandt T (2007) Episodic ataxia type 2. Neurotherapeutics 4: 267-73. doi: 10.1016/j.nurt.2007.01.014
Subramony SH, Schott K, Raike RS, Callahan J, Langford LR, Christova PS, Anderson JH, Gomez CM (2003) Novel CACNA1A mutation causes febrile episodic ataxia with interictal cerebellar deficits. Ann Neurol 54: 725-31. doi: 10.1002/ana.10756
Tomlinson SE, Tan SV, Burke D, Labrum RW, Haworth A, Gibbons VS, Sweeney MG, Griggs RC, Kullmann DM, Bostock H, Hanna MG (2016) In vivo impact of presynaptic calcium channel dysfunction on motor axons in episodic ataxia type 2. Brain 139: 380-91. doi: 10.1093/brain/awv380
Tomlinson SE, Tan SV, Kullmann DM, Griggs RC, Burke D, Hanna MG, Bostock H (2010) Nerve excitability studies characterize Kv1.1 fast potassium channel dysfunction in patients with episodic ataxia type 1. Brain 133: 3530-40. doi: 10.1093/brain/awq318
van den Maagdenberg AM, Kors EE, Brunt ER, van Paesschen W, Pascual J, Ravine D, Keeling S, Vanmolkot KR, Vermeulen FL, Terwindt GM, Haan J, Frants RR, Ferrari MD (2002) Episodic ataxia type 2. Three novel truncating mutations and one novel missense mutation in the CACNA1A gene. J Neurol 249: 1515-9. doi: 10.1007/s00415-002-0860-8
VanDyke DH, Griggs RC, Murphy MJ, Goldstein MN (1975) Hereditary myokymia and periodic ataxia. J Neurol Sci 25: 109-18. doi: 10.1016/0022-510x(75)90191-4
Verriello L, Carrera P, Pauletto G, Bernardini A, Valente M, Gigli GL (2021a) Case report and ten-year follow-up of episodic ataxia type 2 due to a novel variant in CACNA1A. eNeurologicalSci 23: 100334. doi: 10.1016/j.ensci.2021.100334
Verriello L, Pauletto G, Nilo A, Lonigro I, Betto E, Valente M, Curcio F, Gigli GL (2021b) Epilepsy and episodic ataxia type 2: family study and review of the literature. J Neurol 268: 4296-4302. doi: 10.1007/s00415-021-10555-0
von Brederlow B, Hahn AF, Koopman WJ, Ebers GC, Bulman DE (1995) Mapping the gene for acetazolamide responsive hereditary paryoxysmal cerebellar ataxia to chromosome 19p. Hum Mol Genet 4: 279-84. doi: 10.1093/hmg/4.2.279
Wan J, Carr JR, Baloh RW, Jen JC (2005) Nonconsensus intronic mutations cause episodic ataxia. Ann Neurol 57: 131-5. doi: 10.1002/ana.20343
Wan J, Mamsa H, Johnston JL, Spriggs EL, Singer HS, Zee DS, Al-Bayati AR, Baloh RW, Jen JC, Investigators C (2011) Large Genomic Deletions in CACNA1A Cause Episodic Ataxia Type 2. Front Neurol 2: 51. doi: 10.3389/fneur.2011.00051
Xu Y, Wang Z, Sun Q, Zhou L, Xu H, Hu Y (2022) Clinical features and CACNA1A gene mutation in a family with episodic ataxia type 2. Zhong Nan Da Xue Xue Bao Yi Xue Ban 47: 801-808. doi: 10.11817/j.issn.1672-7347.2022.210650
Ying SH, Jen JC, Baloh RW (2001) Similar oculomotor phenotypes in episodic ataxia type 2 and spinocerebellar atrophy type 6. Ann N Y Acad Sci 942: 508-9. doi: 10.1111/j.1749-6632.2001.tb03783.x
Yuan H, Yuan H, Wang Q, Ye W, Yao R, Xu W, Liu Y (2020) Two novel KCNA1 variants identified in two unrelated Chinese families affected by episodic ataxia type 1 and neurodevelopmental disorders. Mol Genet Genomic Med 8: e1434. doi: 10.1002/mgg3.1434
Yuan X, Zheng Y, Gao F, Sun W, Wang Z, Zhao G (2022) Case Report: A Novel CACNA1A Mutation Caused Flunarizine-Responsive Type 2 Episodic Ataxia and Hemiplegic Migraine With Abnormal MRI of Cerebral White Matter. Front Neurol 13: 899813. doi: 10.3389/fneur.2022.899813
Zafeiriou DI, Lehmann-Horn F, Vargiami E, Teflioudi E, Ververi A, Jurkat-Rott K (2009) Episodic ataxia type 2 showing ictal hyperhidrosis with hypothermia and interictal chronic diarrhea due to a novel CACNA1A mutation. Eur J Paediatr Neurol 13: 191-3. doi: 10.1016/j.ejpn.2008.02.011
Zhao J, Petitjean D, Haddad GA, Batulan Z, Blunck R (2020) A Common Kinetic Property of Mutations Linked to Episodic Ataxia Type 1 Studied in the Shaker Kv Channel. Int J Mol Sci 21. doi: 10.3390/ijms21207602
Zhu J, Alsaber R, Zhao J, Ribeiro-Hurley E, Thornhill WB (2012) Characterization of the Kv1.1 I262T and S342I mutations associated with episodic ataxia 1 with distinct phenotypes. Arch Biochem Biophys 524: 99-105. doi: 10.1016/j.abb.2012.05.006
![]()