By Marcello Cherchi, MD PhD
For patients
Guillain-Barré syndrome (GBS) is one of a group of acute inflammatory demyelinating polyradiculoneuropathies (AIDPs). These diseases damage nerves controlling movement, touch, and sometimes nerves in the head, including nerves for balance and eye movements. These diseases are dangerous because they can make it difficult to breathe. After the severe symptoms get better, some patients notice poor balance. In such cases, your doctor may check some tests of hearing and balance, and depending on those results, it might be worth going to a physical therapist to try vestibular rehabilitation therapy (VRT).
For clinicians
Overview
Guillain-Barré syndrome (GBS) is one of a group of acute inflammatory demyelinating polyradiculoneuropathies (AIDPs). GBS was classically described as manifesting clinically with acute symmetrical ascending paralysis; electrophysiologically with peripheral demyelination; and with cerebrospinal fluid findings of elevated albumin without pleocytosis. Other AIDPs exhibit other features (e.g., somatosensory deficits, cranial nerve deficits, autonomic deficits, axonal damage), and some of these diseases have been associated with specific antibodies against various ganglioside proteins (such as GQ1b, GM2 and others). GBS is reported to have an annual incidence of 1 to 2 per 100,000 people, and up to two-thirds of patients report an antecedent infection in the weeks prior to the onset of GBS symptoms. Impaired somatomotor function (weakness) and somatosensory function (afferent deficits) can cause ataxia, but in some cases a patient’s disequilibrium appears to exceed what would be expected from such deficits, and this is the usual reason for referral to otoneurology or neuro-otology. In such patients, if vestibular testing (vestibular evoked myogenic potentials, video head impulse testing, caloric testing) identifies vestibular weakness, then it is medically reasonable to consider a trial of vestibular rehabilitation therapy (even though no systematic trials of such intervention have been conducted), as this incurs no medical risk.
Introduction
Guillain-Barré syndrome (GBS) is named after Georges Charles Guillain (1876 – 1961) and Jean-Alexandre Barré (1880 – 1967), though their original paper was co-authored with André Strohl (1887 – 1977) (Guillain et al. 1916). However, earlier clinical descriptions of this syndrome were published in 1859 (Landry 1859) by Jean Baptiste Octave Landry (1826 – 1865) (Skalski et al. 2019a, b), and probably by many others (Mathis et al. 2020), though designations such as “Landry-Guillain-Barré-Strohl syndrome” (Afifi 1994; Samantray et al. 1977) never caught on.
In the 20th century GBS was regarded as manifesting clinically with acute symmetrical ascending paralysis; electrophysiologically with peripheral demyelination; and with cerebrospinal fluid findings of elevated albumin without pleocytosis.
Subsequent research reported variants of GBS, such as “GBS with sensory findings,” “GBS with autonomic dysfunction,” “GBS with axonal neuropathy,” and eventually also various associated autoantibody patterns. In view of this variety, GBS has been subsumed under a group of acute immune-mediated neuropathies referred to as acute inflammatory demyelinating polyradiculoneuropathy (AIDP).
Two “subtypes of GBS” — Miller Fisher syndrome (Fisher 1956) and Bickerstaff brainstem encephalitis (Bickerstaff and Cloake 1951) — ultimately were found to have the pathological mechanism of anti-GQ1b ganglioside autoantibodies (Chiba et al. 1992). Variants with other anti-ganglioside antibodies have also been reported (Yuki and Hartung 2012).
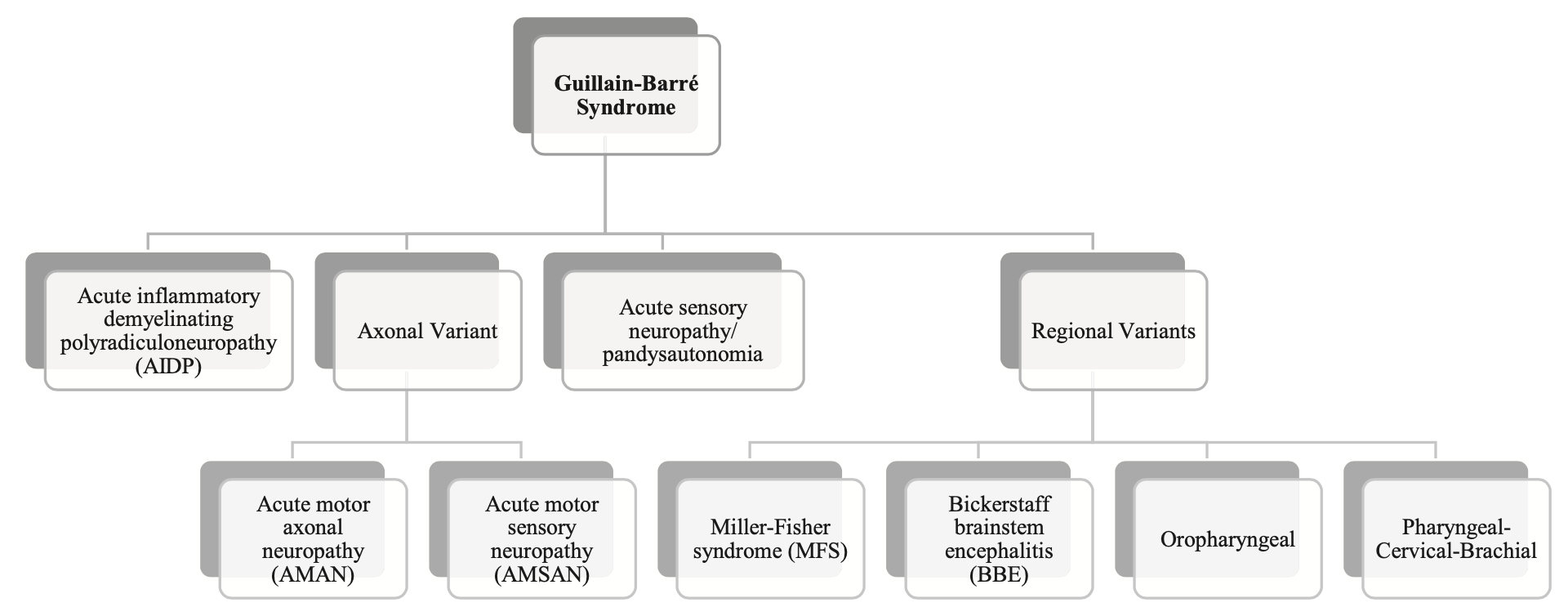
Wanleenuwat and colleagues proposed the taxonomy of AIDP shown in the Figure below.
Epidemiology
The annual incidence of GBS is reported as 1 to 2 cases per 100,000 people (McGrogan et al. 2009; Sejvar et al. 2011; Shahrizaila et al. 2021; Yuki and Hartung 2012).
Genetics
It is not known whether genetic factors modulate the risk of developing GBS.
Pathophysiological mechanism of disease
Up to two thirds of patients ultimately diagnosed with GBS report an antecedent infection in the weeks prior to onset of neuromuscular symptoms. It is thought that epitopes shared between the pathogen and native proteins trigger a cross-reactive immune response, referred to as molecular mimicry. A range of antecedent infections has been reported, including bacterial (Campylobacter jejuni, Hemophilus influenza, Escherichia coli, Mycobacterium pneumonia) and viral (cytomegalovirus, influenza A and B, HIV, COVID-19, Zika, varicella-zoster, Epstein-Barr, herpes simplex and Japanese encephalitis virus). GBS has also been reported following some vaccinations.
GBS and other AIDPs can interfere with a person’s equilibrium through several mechanisms, including:
- Motoric impairment (muscle weakness) will interfere with the support required for maintaining an upright posture and ambulation.
- For the less common variants of AIDP that include sensory involvement, this afferent defect can interfere with equilibrium similar to other sensory neuropathies.
- Peripheral vestibular weakness, or impairment of central vestibular function (more on these below).
- If AIDP interferes with ocular motor function (such as Miller Fisher syndrome), deficits such as diplopia can interfere with stereopsis and thus with depth perception, contributing to disequilibrium.
Clinical presentation
Blanquet and colleagues (Blanquet et al. 2018) studied 5 GBS patients. Of those 5 patients they reported that one (20%) had neither vestibular symptoms nor hearing loss; one (20%) had both vestibular symptoms and hearing loss; one (20%) had hearing loss but no vestibular symptoms; and two (40%) had vestibular symptoms but no hearing loss.
Physical examination
Patients with AIDP exhibit weakness and often (but not always) diminished myotatic reflexes.
Ocular motor examination
Patients with Miller Fisher syndrome exhibit varying degrees of ophthalmoplegia. Patients with anti-GQ1b antibodies can exhibit spontaneous down beat nystagmus, periodic alternating nystagmus, gaze-evoked nystagmus, central positional nystagmus, ocular flutter and saccadic dysmetria (Lee et al. 2024).
Testing: vestibular
Blanquet and colleagues (Blanquet et al. 2018) studied 5 GBS patients with video head impulse testing (vHIT) and vestibular evoked myogenic potentials (VEMPs). Of these 5 GBS patients, four (80%) exhibited some abnormality on vestibular testing. In one GBS patient (20%) cervical vestibular evoked myogenic potentials (cVEMP) exhibited unilaterally prolonged p13 and n23 latencies; the remainder had normal cVEMPs. Two GBS patients (40%) exhibited unilaterally prolonged p15 latencies on ocular vestibular evoked myogenic potentials (oVEMP); the remainder had normal oVEMPs. Two GBS patients (40%) exhibited abnormal video head impulse testing (vHIT) in at least one semicircular canal.
Lee and colleagues (Lee et al. 2019) studied anti-GQ1b antibodies in patients presenting with acute vestibular syndrome, and built on that series in a later study as well (Lee et al. 2024). They studied patients with GQ1b antibodies. They note that:
“GQ1b gangliosides are… present in a high concentration in the vestibulocochlear nerves. Therefore, canal paresis can be observed during bithermal caloric tests in patients with MFS [Miller Fisher syndrome] but without ophthalmoplegia. Furthermore, typical features of acute unilateral peripheral vestibulopathy (i.e., vestibular neuritis) may be observed in association with serum anti-GQ1b antibody positivity” (Lee et al. 2024).
They specifically reported anti-GQ1b antibodies in 12 patients with acute unilateral peripheral vestibulopathy, commenting that, “In these cases, abnormal head-impulse test results and canal paresis on bithermal caloric tests mostly indicate peripheral vestibulopathy” (Lee et al. 2024). However, they also noted that, “Spontaneous downbeat nystagmus, periodic alternating [nystagmus], gaze-evoked [nystagmus], and central positional nystagmus; ocular flutter; and saccadic dysmetria have been described in association with anti-GQ1b antibody positivity” (Lee et al. 2024), and interpreted these as central findings. They therefore concluded that, “Anti-GQ1b antibody syndrome should be considered in patients presenting with either peripheral or central vestibulopathy of a probable immune-mediated etiology” (Lee et al. 2024).
Kim and colleagues (Kim et al. 2018) studied 8 patients who had a GBS-like syndrome and were positive for either IgG, or IgM, or IgG plus IgM anti-GM2-ganglioside antibodies (and negative for other antibodies such as GQ1b). They reported that of the four patients who were IgG positive, all (100%) complained of dizziness and three (75%) complained of ataxia. All four patients (100%) exhibited involvement of more than one cranial nerve; two (50%) had oculomotor nerve involvement, one (25%) had abducens nerve involvement; and one (25%) had “probable involvement of the vestibular nerve.”
Testing: other
Electromyography with nerve conduction velocities (EMG/NCV) may show specific features supportive of a diagnosis of AIDP.
Cerebrospinal fluid analysis shows elevated protein but a normal white cell count.
Imaging
MRI may show enhancement of affected cranial nerves or spinal nerve roots.
Histopathology
The histopathology of GBS has been described in detail (Asbury et al. 1969), and is usually characterized by “demyelination and multifocal perivascular and endoneurial T-cell infiltrations with patchy involvement of spinal roots and nerve trunks and distal nerve segments” (Sheikh 2020). Some investigations suggest that the damage to Schwann cells is complement-mediated (Hafer-Macko et al. 1996).
Differential diagnosis
Shaikh (Sheikh 2020) offers the following differential diagnostic considerations for GBS:
- Inflammatory/immune
- Acute-onset chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) Note that CIDP is not simply the “chronic phase of AIDP,” rather, CIDP is a different disease.
- Vasculitic neuropathy
- Sensory ganglionitis
- Intensive care unit setting
- Critical illness neuropathy
- Metabolic
- Diabetes
- Porphyria
- Nutritional
- Thiamine deficiency
- Infectious
- Human immunodeficiency virus (HIV)
- Herpes viruses
- Lyme disease
- Toxic
- Organophosphates
- Hexacarbons
Treatment
GBS and other variants of AIDP require hospitalization to stabilize respiratory and autonomic function. Inpatient treatment with immunotherapies such as plasma exchange and intravenous immune globulin are often used.
Otoneurologists and neuro-otologists are generally not the first practitioners to diagnose AIDP. In our experience, the more common scenario is that an individual already diagnosed with AIDP has partially recovered but is experiencing disequilibrium beyond what would be expected from neuromuscular weakness alone, and this is the usual reason for referral.
In such cases it is reasonable to check tests of peripheral vestibular function (cervical and ocular vestibular evoked myogenic potentials, video head impulse testing, videonystagmography and rotatory chair testing). If vestibular weakness (symmetrical or asymmetrical) is identified, then referral to vestibular rehabilitation therapy (VRT) is reasonable (though this has not been studied in controlled trials) and incurs no medical risk.
We have not personally encountered AIDP patients with clinically significant hearing loss that was confidently attributable to the AIDP. In such cases, it would be reasonable to consult audiology to be evaluated for amplification.
Prognosis
GBS and other variants of AIDP can be life-threatening when they impair respiratory function or autonomic dysfunction.
As far as vestibular and auditory symptoms are concerned, studies regarding the natural history and prognosis are lacking.
References
Afifi AK (1994) The Landry-Guillain-Barre Strohl syndrome 1859 to 1992 a historical perspective. J Family Community Med 1: 30-4.
Asbury AK, Arnason BG, Adams RD (1969) The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine (Baltimore) 48: 173-215. doi: 10.1097/00005792-196905000-00001
Bickerstaff ER, Cloake PC (1951) Mesencephalitis and rhombencephalitis. Br Med J 2: 77-81. doi: 10.1136/bmj.2.4723.77
Blanquet M, Petersen JA, Palla A, Veraguth D, Weber KP, Straumann D, Tarnutzer AA, Jung HH (2018) Vestibulo-cochlear function in inflammatory neuropathies. Clin Neurophysiol 129: 863-873. doi: 10.1016/j.clinph.2017.11.025
Chiba A, Kusunoki S, Shimizu T, Kanazawa I (1992) Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol 31: 677-9. doi: 10.1002/ana.410310619
Fisher M (1956) An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med 255: 57-65. doi: 10.1056/NEJM195607122550201
Guillain G, Barré J-A, Strohl A (1916) Sur un syndrome de radiculo-névrite avec hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire: Remarque sur les caractères cliniques et graphiques des réflexes tendineux [On a syndrome of radiculoneuritis with hyperalbuminosis of the cerebrospinal fluid without a cellular reaction. Remarks on the clinical characteristics and tracings of the tendon reflexes]. Bulletins et Mémoires de la Société Médicale des Hôpitaux de Paris 40: 1462.
Hafer-Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, Asbury AK, Griffin JW (1996) Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol 39: 625-35. doi: 10.1002/ana.410390512
Kim JK, Kim YH, Yoon BA, Cho JY, Oh SY, Shin HY, Kim JS, Park KH, Kim SY, Suh BC, Seok HY, Yoo JH, Bae JS (2018) Clinical Heterogeneity of Anti-GM2-Ganglioside-Antibody Syndrome. J Clin Neurol 14: 401-406. doi: 10.3988/jcn.2018.14.3.401
Landry JBO (1859) Note sur la paralysie ascendante aiguë [Note on acute ascending paralysis]. Gazette hébdomadaire de médecine et de chirurgie 6: 472-474.
Lee SU, Kim HJ, Choi JY, Choi KD, Kim JS (2024) Expanding Clinical Spectrum of Anti-GQ1b Antibody Syndrome: A Review. JAMA Neurol 81: 762-770. doi: 10.1001/jamaneurol.2024.1123
Lee SU, Kim HJ, Choi JY, Kim JK, Kim JS (2019) Acute vestibular syndrome associated with anti-GQ1b antibody. Neurology 93: e1085-e1092. doi: 10.1212/wnl.0000000000008107
Mathis S, Soulages A, Vallat JM, Le Masson G (2020) History of acute polyradiculoneuropathy (part 1): The prehistory of Guillain-Barre syndrome. Neurology 94: 828-835. doi: 10.1212/WNL.0000000000009401
McGrogan A, Madle GC, Seaman HE, de Vries CS (2009) The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology 32: 150-63. doi: 10.1159/000184748
Samantray SK, Johnson SC, Mathai KV, Pulimood BM (1977) Landry-Guillain-Barre-Strohl syndrome. A study of 302 cases. Med J Aust 2: 84-91. doi: 10.5694/j.1326-5377.1977.tb99056.x
Sejvar JJ, Baughman AL, Wise M, Morgan OW (2011) Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology 36: 123-33. doi: 10.1159/000324710
Shahrizaila N, Lehmann HC, Kuwabara S (2021) Guillain-Barre syndrome. Lancet 397: 1214-1228. doi: 10.1016/S0140-6736(21)00517-1
Sheikh KA (2020) Guillain-Barre Syndrome. Continuum (Minneap Minn) 26: 1184-1204. doi: 10.1212/CON.0000000000000929
Skalski P, Owecki MK, Magowska AM (2019a) Correction to: Jean Baptiste Octave Landry (1826–1865). Journal of Neurology 266: 2344-2344. doi: 10.1007/s00415-018-9152-9
Skalski P, Owecki MK, Magowska AM (2019b) Jean Baptiste Octave Landry (1866–1940). Journal of Neurology 266: 2341-2343. doi: 10.1007/s00415-018-9120-4
Wanleenuwat P, Iwanowski P, Kozubski W (2020) Antiganglioside antibodies in neurological diseases. J Neurol Sci 408: 116576. doi: 10.1016/j.jns.2019.116576
Yuki N, Hartung HP (2012) Guillain-Barre syndrome. N Engl J Med 366: 2294-304. doi: 10.1056/NEJMra1114525
![]()