By Marcello Cherchi, MD PhD
For patients
Friedreich ataxia (FA) is a genetic cause of ataxia. It usually begins slowly in childhood or teenage years, typically with clumsiness, falls, and difficulty pronouncing words, and these symptoms gradually worsen. Other problems include high arched feet, abnormal curvature of the spine, and abnormal thickening of the walls of the heart. Your doctor may suspect this disease based on your clinical history and physical examination; other tools that can be helpful in diagnosis include a brain MRI and genetic testing. There is no curative treatment for this disease, so management usually involves care to make a patient safe and comfortable.
For clinicians
Overview
Friedreich ataxia is the most common hereditary ataxia, transmitted in an autosomal recessive fashion. The genetic abnormality is a trinucleotide repeat expansion in the frataxin gene on chromosome 9q13 that encodes for an iron trafficking protein in mitochondria, malfunction of which leads to the production of free radicals resulting in oxidative stress and cell death. The greater the number of trinucleotide repeats, the more severe and more rapidly progressive the disease. The disease usually begins insidiously in childhood or young adulthood, and the initial symptoms usually include gait ataxia and dysarthria. Aside from the prominent abnormalities in the nervous system, other organ systems are also affected, including cardiovascular (hypertrophic cardiomyopathy), endocrine (diabetes) and skeletal (pes cavus, scoliosis). Physical examination usually shows appendicular ataxia and weakness, cerebellar dysarthria, reduced proprioception and vibratory sensation, and the skeletal abnormalities mentioned before. Ocular motor examination may show eye movements compatible with cerebellar dysfunction, including spontaneous nystagmus, saccadic dysmetria and poor smooth pursuit. Audiometry may often shows hearing loss and abnormal brainstem auditory evoked responses. Instrumented vestibular testing usually shows a variety of abnormalities as well. Brain MRI may show atrophy of the superior cerebellar vermis. No pharmacotherapy has yet been shown to alter the course of the disease, so management is symptomatic, and usually involves multiple specialists (neurology, cardiology, endocrinology, speech pathology, occupational and physical therapy, and medical genetics).
Introduction
Friedreich ataxia (FA) is the most common hereditary ataxia, and is transmitted in an autosomal recessive fashion.
The disease was first described by the German neuropathologist Nikolaus Friedreich (1825 – 1922) in 1863 (Friedreich 1863).

Epidemiology
Friedreich ataxia is the most common hereditary ataxia in the Caucasian population (Wallace and Bird 2018). The estimated prevalence of FA is 1 in 50,000, with a carrier frequency in the Caucasian population of 1 in 90 (Cossee, Durr et al. 1999).
Genetics
Friedreich ataxia (OMIM 229300) results from a trinucleotide (GAA) repeat expansion in the frataxin gene located at chromosome 9q21.11 (OMIM 606829). This abnormality is found in the first intron of the gene in 98% of individuals (Cossee, Durr et al. 1999). There is an inverse relationship between the length of the trinucleotide repeat expansion and the onset/severity of symptoms.
Pathophysiological mechanism of disease
The genetic mutation of this disease affects frataxin, a nuclear-encoded protein expressed at the inner mitochondrial membrane that is involved in the influx/efflux of iron into and out of mitochondria. It is suspected that this faulty iron metabolism results in elevated levels of free radicals, leading to mitochondrial damage and eventually cell death (Cossee, Durr et al. 1999).
Clinical presentation
The usual age of onset is between 5 – 15 years of age (Cossee, Durr et al. 1999), though late-onset cases have been reported. Durr and colleagues (Durr, Cossee et al. 1996) studied 140 FA patients and reported the age of onset to range from 2 to 51 years (mean 15.5±8 years).
The most common symptoms include ataxia, clumsiness and dysarthria (Cossee, Durr et al. 1999). In the series of 140 FA patients studied by Durr and colleagues (Durr, Cossee et al. 1996) the authors state, “The presenting symptom was gait ataxia in all patients except for several in whom scoliosis was diagnosed before the ataxia.” The meta analysis of 650 genetically confirmed FA patients by Reetz and colleagues (Reetz, Dogan et al. 2018) reported that the most common presenting symptoms were instability (77.2%), scoliosis (23.1%) and falls (19.8%). Further details are displayed in the Table below.
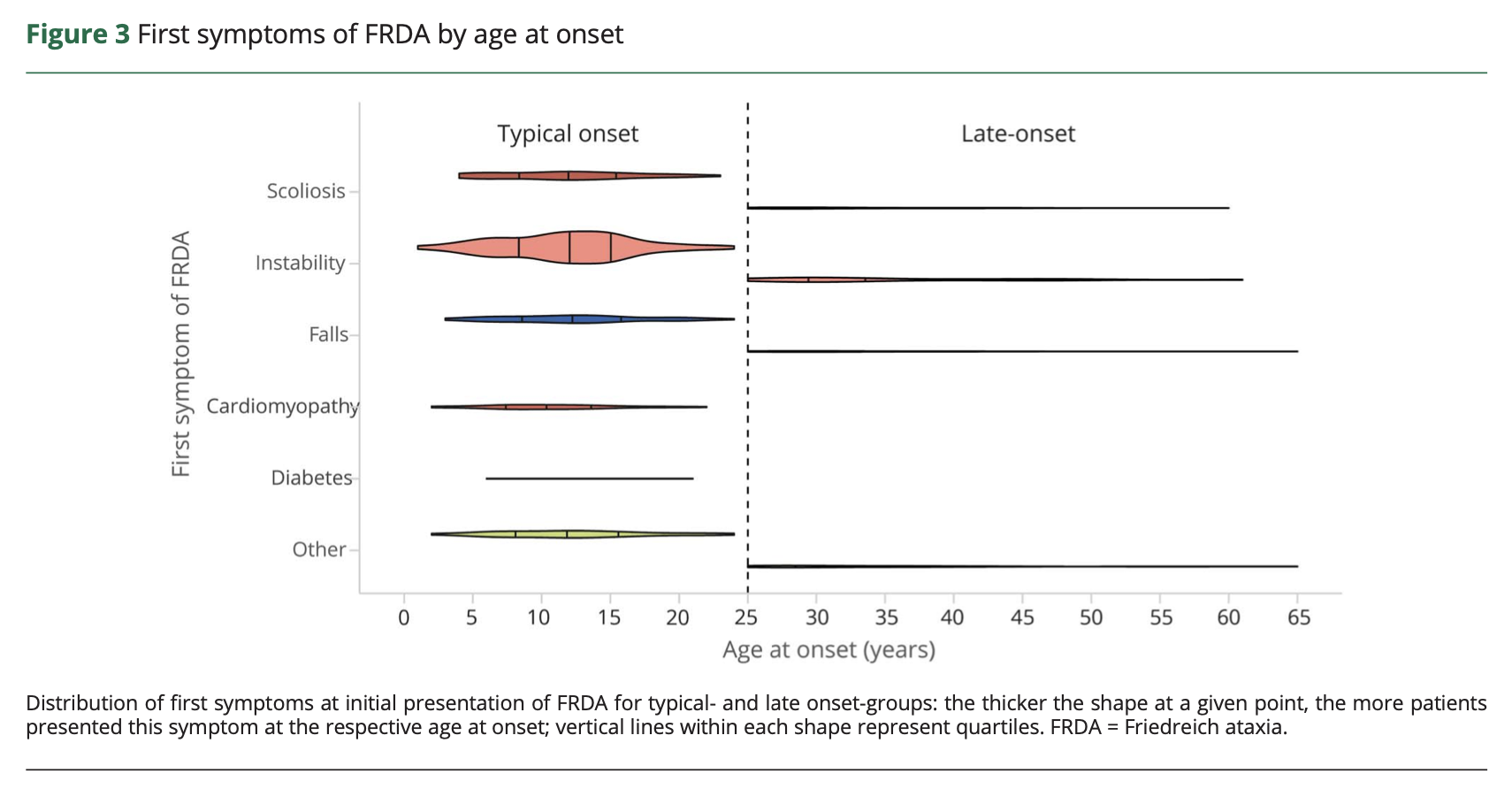
The Table below outlines the clinical features in 140 FA patients studied by Durr and colleagues (Durr, Cossee et al. 1996).
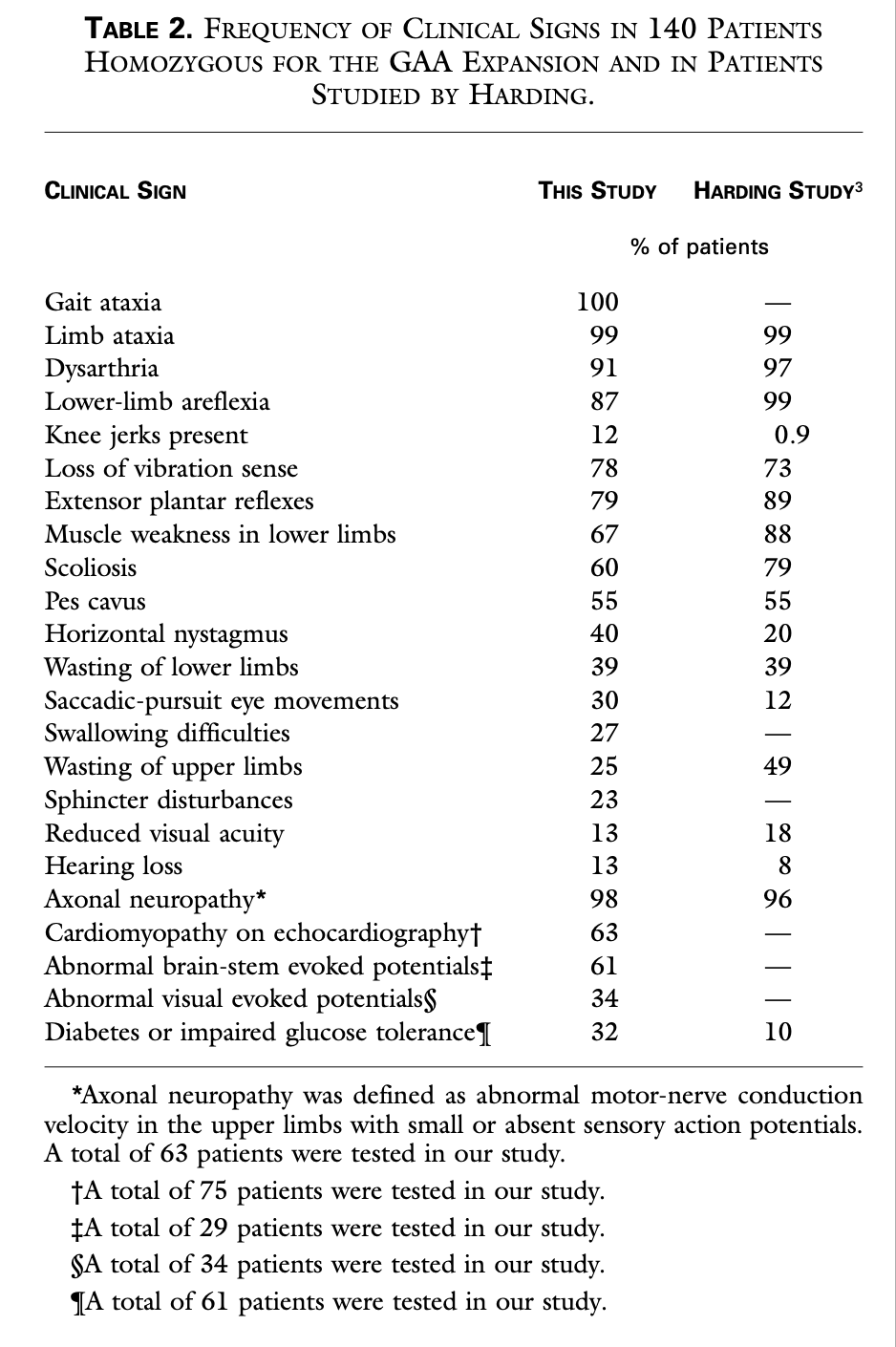
There is variability in clinical presentation, some of which appears to be correlated with the specific pattern of genetic mutation (Cossee, Durr et al. 1999). The later onset cases appear to be related to a more limited number of trinucleotide repeats in the abnormal frataxin gene.
The Table below, from Durr and colleagues (Durr, Cossee et al. 1996), shows that a higher number of trinucleotide repeats correlates with earlier mean age of onset, shorter time to becoming wheelchair-bound, greater prominence of physical examination abnormalities, greater frequency of diabetes and cardiomyopathy.
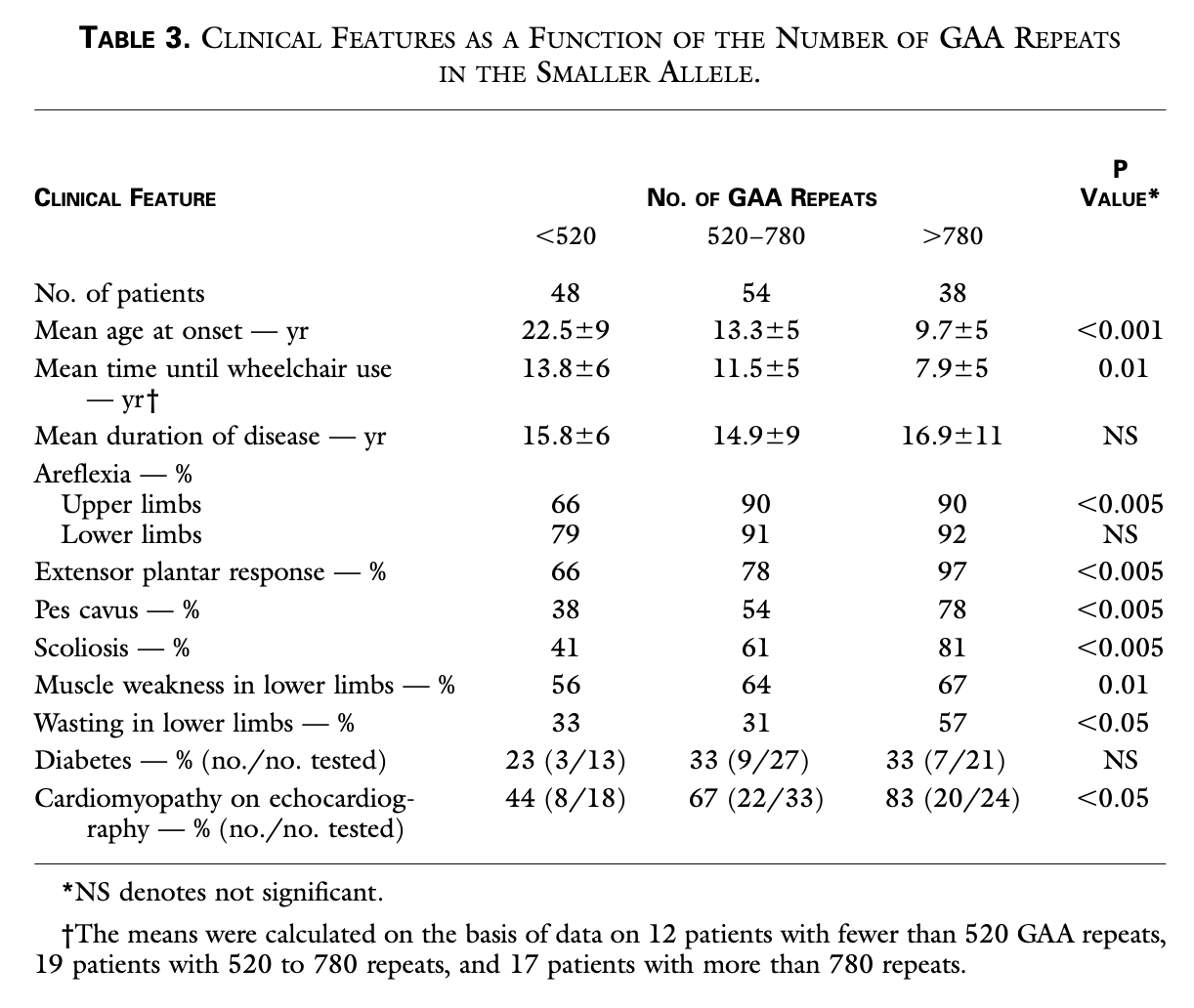
Reetz and colleagues (Reetz, Dogan et al. 2018) present a meta analysis of studies including 650 genetically confirmed FA patients. They describe in detail the non-ataxia symptoms in FA and report that, “The most frequent clinical features beyond afferent ataxia were abnormal eye movements (90.5%), scoliosis (73.5%), deformities of the feet (58.8%), urinary dysfunction (42.8%), cardiomyopathy and cardiac hypertrophy (40.3%), followed by decreased visual acuity (36.8%); less frequent features were, among others, depression (14.1%) and diabetes (7.1%).”
Physical examination
Physical examination usually shows appendicular ataxia, muscle weakness, reduced or absent myotatic reflexes, present Babinski signs, reduced or absent vibratory sensation in the lower limbs, and dysarthria in a cerebellar pattern (Cossee, Durr et al. 1999).
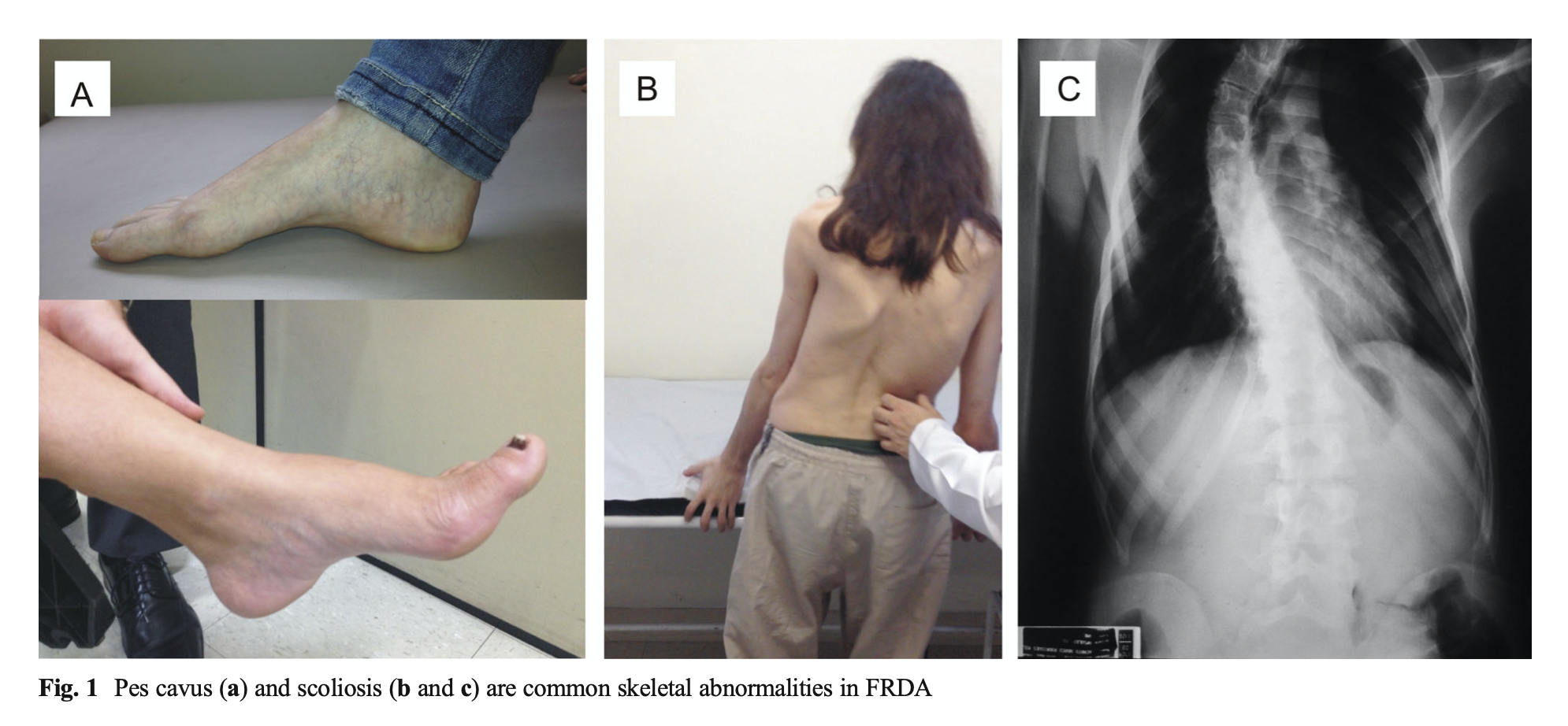
Patients also develop skeletal abnormalities including scoliosis and pes cavus, as shown in the Figure below from Abrahao and colleagues (Abrahao, Pedroso et al. 2015).
Ocular motor examination
Ocular motor abnormalities are common in FA. On face-to-face examination, or with infrared video Frenzel goggles, one may find spontaneous nystagmus, saccadic dysmetria and poor smooth pursuit (all of which suggest cerebellar localization).
The Table below summarizes the ocular motor findings and some vestibular test result abnormalities of 30 genetically confirmed FA patients reported by Ziegelboim and colleagues (Zeigelboim, Mesti et al. 2017).
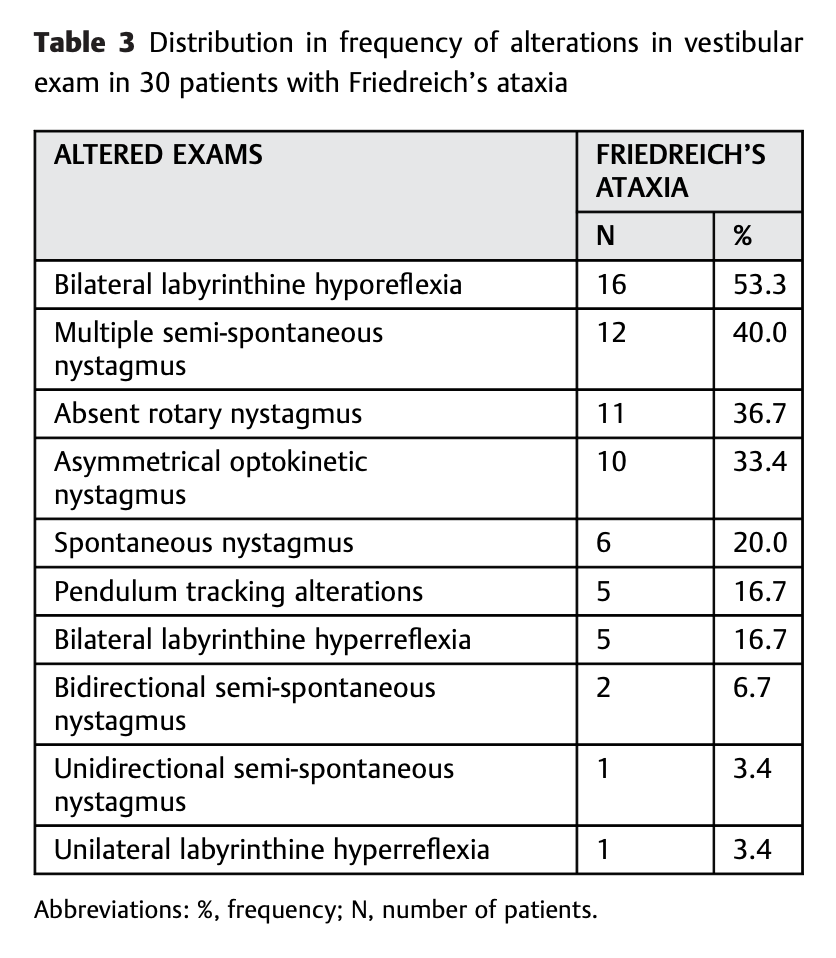
Testing: auditory
FA patients generally do not complain of hearing loss. Nevertheless, the majority have abnormalities on hearing tests.
Ell and colleagues (Ell, Prasher et al. 1984) studied 10 patients with genetically confirmed FA. All 10 patients underwent audiometry, of which 9 (90%) were abnormal. The Figure below shows a composite audiogram comparing the mean hearing thresholds of FA patients with “neurological control subjects.”
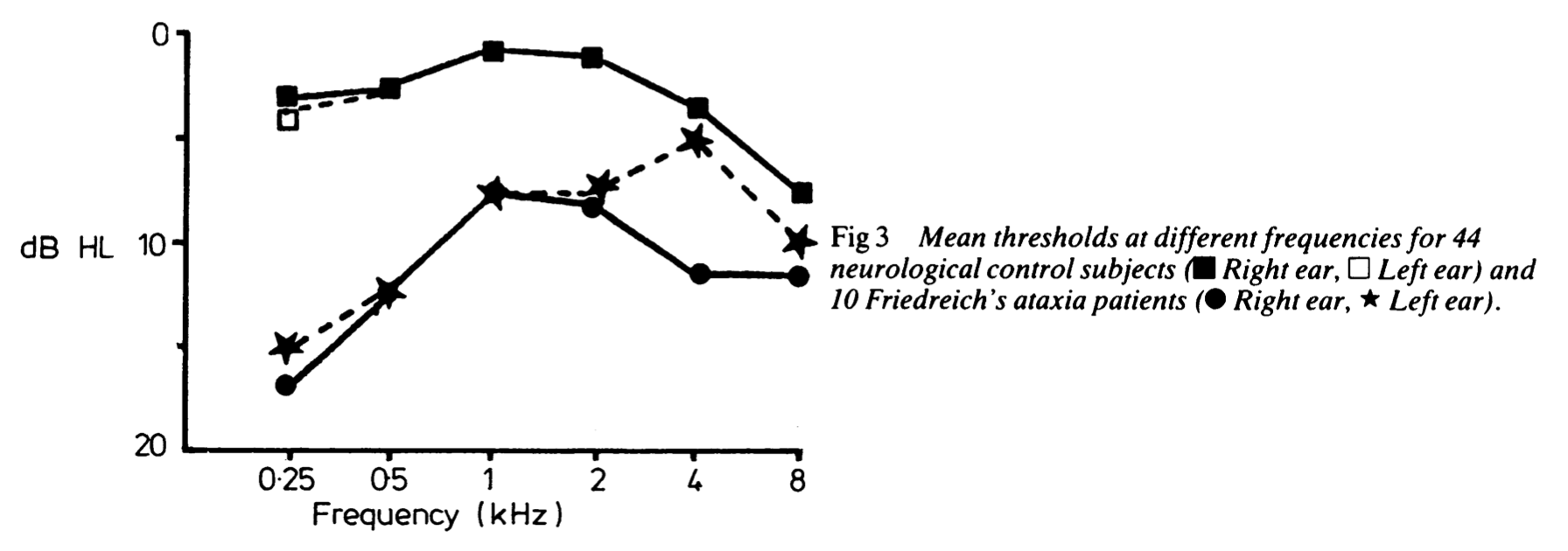
We find it striking that the hearing loss is more pronounced in the low frequencies; this suggests greater pathology at the cochlear apex or its afferents.
Ell and colleagues (Ell, Prasher et al. 1984) studied auditory evoked brainstem responses (ABRs) in 7 genetically confirmed FA patients and reported that 6 (86%) were abnormal.
Testing: vestibular
The majority of FA patients exhibit ocular motor abnormalities. The Table below compiles the statistics reported in several studies (Baloh et al. 1975; Ell et al. 1984; Fahey et al. 2008; Furman et al. 1983; Harding 1981; Kirkham et al. 1979; Luis et al. 2016; Ribai et al. 2007).
|
Fahey et al. 2008 |
Kirkham et al. 1979 |
Ell et al. 1984 |
Furman et al. 1983 |
Baloh et al. 1975 |
Harding et al. 1981 |
Luis et al. 2016 |
Ribal et al 2007 |
|
|
Saccades: dysmetria |
71% |
88% |
||||||
|
Saccades: hypermetria |
37% |
|||||||
|
Saccades: hypometria |
9% |
|||||||
|
Saccades: latency prolonged |
100% |
|||||||
|
Smooth pursuit: abnormal |
100% |
86% |
90% |
100% |
12.2% |
|||
|
Nystagmus: spontaneous down beat |
45% |
|||||||
|
Nystagmus: horizontal |
80% |
|||||||
|
Nystagmus: rebound |
57% |
50% |
||||||
|
Nystagmus: gaze-evoked |
80% |
|||||||
|
Nystagmus: gaze paretic |
64% |
|||||||
|
Nystagmus: gaze evoked paretic, horizontal |
60% |
80% |
||||||
|
Nystagmus: gaze-evoked paretic, vertical |
20% |
|||||||
|
“Nystagmus” (not further specified) |
20% |
|||||||
|
Square wave jerks |
43% |
70% |
94% |
|||||
|
“Fixation instability” |
100% |
|||||||
|
Flutter |
50% |
|||||||
|
Optokinetic responses: abnormal |
43% |
20% |
||||||
|
Caloric responses reduced or absent |
100% |
100% |
||||||
|
vHIT: reduced VOR gain |
100% |
100% |
||||||
|
vHIT: overt saccades present |
89% |
|||||||
|
VOR suppression: abnormal |
86% |
60% |
||||||
|
RCT: reduced VOR gain |
31% |
|||||||
|
RCT: reduced VVOR gain |
100% |
|||||||
|
RCT: increased VFX gain |
100% |
Some of the outliers in the above table (such as the 12.2% of patients with abnormal smooth pursuit reported by Harding and colleagues) were probably due to methodological differences.
Some studies report results in a continuous rather than dichotomous fashion. For example, Spieker and colleagues (Spieker, Schulz et al. 1995) noted the following statistically significant differences of FA patients compared to controls: reduced saccade velocity, reduced optokinetic gain, reduced vestibulo-ocular reflex gain, reduced time constant, reduced vestibulo-ocular reflex suppression and reduced total caloric response.
There are also reports of FA patients exhibiting periodic alternating nystagmus (Gorman and Brock 1950, Gorman, Brock et al. 1950).
In summary, the most common ocular motor abnormalities observed in FA include saccadic dysmetria, prolonged saccadic latencies, abnormal smooth pursuit, abnormal nystagmus (spontaneous, rebound) and square wave jerks. Instrumented vestibular testing shows abnormalities on caloric testing (reduced responses), video head impulse testing (reduced vestibulo-ocular reflex gain, presence of overt compensatory saccades) and rotatory chair testing (reduced visual vestibulo-ocular reflex gain, increased visual fixation suppression gain).
Testing: other
Patients invariably develop cardiac abnormalities including electrocardiographic or echocardiographic signs of hypertrophic cardiomyopathy.
About 30% of patients develop diabetes (Cossee, Durr et al. 1999).
Imaging
Brain MRI often shows cerebellar atrophy, particularly in the superior vermis (Hadjivassiliou, Wallis et al. 2012).
Histopathology
Autopsy studies of patients with FA (Jitpimolmard, Small et al. 1993) typically show central nervous system findings that include degeneration of the posterior columns, ventral and dorsal spinocerebellar tracts (Lubozynski and Roelofs 1975), loss of dorsal root ganglion cells, loss of neurons in the nucleus dorsalis of the spinal cord, the spinal and principal trigeminal nuclei and the mesencephalic trigeminal nucleus. Peripheral nervous system findings include distal axonal peripheral neuropathy (tending to affect larger myelinated fibers that transmit proprioception and vibratory sensation).
Differential diagnosis
Other diseases that can (at least initially) resemble FA include ataxia with vitamin E deficiency (Durr, Cossee et al. 1996), ataxia-telangiectasia, Charcot-Marie-Tooth disease, abetalipoproteinemia and Refsum disease.
Treatment
Given the suspicion that the mechanism of disease involves free radical production due to abnormal iron trafficking in mitochondria (Cossee, Durr et al. 1999), it is logical to explore interventions such as antioxidant therapies (Barca, Emmanuele et al. 2019).
Idebenone, a synthetic analogue of ubiquinone (coenzyme Q10), is an anti-oxidant that may improve performance of the electron transport chain in mitochondria.
Despite this logical therapeutic target, neurological outcomes of treatment with idebenone have been disappointing. A phase 3 prospective, randomized, double-blinded, placebo-controlled trial of idebenone over 6 months showed no different in neurological outcome in the drug versus placebo (Lynch, Perlman et al. 2010). A placebo-controlled study of idebenone found no statistically significant difference in patient perception of symptomatic benefit (Cook, Boesch et al. 2019).
Cardiac outcomes of treatment with idebenone may be different. For example, a prospective, randomized, placebo-controlled trial of idebenone reported “significant reductions of interventricular septal wall thickness and left ventricular mass in the idebenone group versus the placebo group” (Mariotti, Solari et al. 2003).
One prospective, open-label, single-arm trial combined idebenone with the iron chelator, deferiprone, and reported that the combination brought about a “stabilizing effect in neurologic dysfunctions due to an improvement in the kinetic functions, [but] with a worsening of gait and posture scores. Heart hypertrophy parameters and iron deposits in dentate nucleus improved significantly” (Velasco-Sanchez, Aracil et al. 2011). Given that this was an open-label study, we can draw no confident conclusions from these results.
Some experimental work suggested that interferon gamma 1b may increase frataxin levels, but a placebo-controlled trial of this drug found no difference in outcomes (Lynch, Hauser et al. 2019).
Since there is no therapy to retard, arrest or reverse the underlying pathology, management usually focuses on symptomatic management. Given the multi-faceted nature of FA, management usually involves multiple subspecialties, including a neurologist (if possible, an ataxia specialist), cardiology, endocrinology (to monitor for and treat glucose intolerance or diabetes), speech pathology (for swallowing evaluations), occupational and physical therapy, and medical genetics.
Prognosis
The disease is relentlessly progressive, eventually leading to inability to ambulate, leaving a patient wheelchair-bound.
Survival in FA is usually limited by cardiac complications, the degree of which appears dependent on the extent of the trinucleotide repeat expansion (Pousset, Legrand et al. 2015). In the series of 133 genetically confirmed FA patients studied by Pousset and colleagues (Pousset, Legrand et al. 2015), the cause of death in 80% was cardiac in origin, usually from heart failure.
References
Abrahao A, Pedroso JL, Braga-Neto P, Bor-Seng-Shu E, de Carvalho Aguiar P, Barsottini OG (2015) Milestones in Friedreich ataxia: more than a century and still learning. Neurogenetics 16: 151-60. doi: 10.1007/s10048-015-0439-z
Baloh RW, Konrad HR, Honrubia V (1975) Vestibulo-ocular function in patients with cerebellar atrophy. Neurology 25: 160-8. doi: 10.1212/wnl.25.2.160
Barca E, Emmanuele V, DiMauro S, Toscano A, Quinzii CM (2019) Anti-Oxidant Drugs: Novelties and Clinical Implications in Cerebellar Ataxias. Curr Neuropharmacol 17: 21-32. doi: 10.2174/1570159X15666171109125643
Cook A, Boesch S, Heck S, Brunt E, Klockgether T, Schols L, Schulz A, Giunti P (2019) Patient-reported outcomes in Friedreich’s ataxia after withdrawal from idebenone. Acta Neurol Scand 139: 533-539. doi: 10.1111/ane.13088
Cossee M, Durr A, Schmitt M, Dahl N, Trouillas P, Allinson P, Kostrzewa M, Nivelon-Chevallier A, Gustavson KH, Kohlschutter A, Muller U, Mandel JL, Brice A, Koenig M, Cavalcanti F, Tammaro A, De Michele G, Filla A, Cocozza S, Labuda M, Montermini L, Poirier J, Pandolfo M (1999) Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol 45: 200-6. doi: 10.1002/1531-8249(199902)45:2<200::aid-ana10>3.0.co;2-u
Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335: 1169-75. doi: 10.1056/NEJM199610173351601
Ell J, Prasher D, Rudge P (1984) Neuro-otological abnormalities in Friedreich’s ataxia. J Neurol Neurosurg Psychiatry 47: 26-32. doi: 10.1136/jnnp.47.1.26
Fahey MC, Cremer PD, Aw ST, Millist L, Todd MJ, White OB, Halmagyi M, Corben LA, Collins V, Churchyard AJ, Tan K, Kowal L, Delatycki MB (2008) Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain 131: 1035-45. doi: 10.1093/brain/awm323
Friedreich N (1863) Ueber degenerative Atrophie der spinalen Hinterstränge. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin 26: 433-459. doi: 10.1007/BF01878006
Furman JM, Perlman S, Baloh RW (1983) Eye movements in Friedreich’s ataxia. Arch Neurol 40: 343-6. doi: 10.1001/archneur.1983.04050060043006
Gorman WF, Brock S (1950) Periodic alternating nystagmus in Friedreich’s ataxia. Am J Ophthalmol 33: 860-4.
Gorman WF, Brock S, Kestenbaum A (1950) Periodic alternating nystagmus in Friedreich’s ataxia. J Nerv Ment Dis 112: 437-9.
Hadjivassiliou M, Wallis LI, Hoggard N, Grunewald RA, Griffiths PD, Wilkinson ID (2012) MR spectroscopy and atrophy in Gluten, Friedreich’s and SCA6 ataxias. Acta Neurol Scand 126: 138-43. doi: 10.1111/j.1600-0404.2011.01620.x
Harding AE (1981) Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104: 589-620. doi: 10.1093/brain/104.3.589
Jitpimolmard S, Small J, King RH, Geddes J, Misra P, McLaughlin J, Muddle JR, Cole M, Harding AE, Thomas PK (1993) The sensory neuropathy of Friedreich’s ataxia: an autopsy study of a case with prolonged survival. Acta Neuropathol 86: 29-35. doi: 10.1007/BF00454895
Kirkham TH, Guitton D, Katsarkas A, Kline LB, Andermann E (1979) Oculomotor abnormalities in Friedreich’s ataxia. Can J Neurol Sci 6: 167-72. doi: 10.1017/s0317167100119584
Lubozynski MF, Roelofs RI (1975) Friedreich’s ataxia. South Med J 68: 757-63. doi: 10.1097/00007611-197506000-00026
Luis L, Costa J, Munoz E, de Carvalho M, Carmona S, Schneider E, Gordon CR, Valls-Sole J (2016) Vestibulo-ocular reflex dynamics with head-impulses discriminates spinocerebellar ataxias types 1, 2 and 3 and Friedreich ataxia. J Vestib Res 26: 327-34. doi: 10.3233/VES-160579
Lynch DR, Hauser L, McCormick A, Wells M, Dong YN, McCormack S, Schadt K, Perlman S, Subramony SH, Mathews KD, Brocht A, Ball J, Perdok R, Grahn A, Vescio T, Sherman JW, Farmer JM (2019) Randomized, double-blind, placebo-controlled study of interferon-gamma 1b in Friedreich Ataxia. Ann Clin Transl Neurol 6: 546-553. doi: 10.1002/acn3.731
Lynch DR, Perlman SL, Meier T (2010) A phase 3, double-blind, placebo-controlled trial of idebenone in friedreich ataxia. Arch Neurol 67: 941-7. doi: 10.1001/archneurol.2010.168
Mariotti C, Solari A, Torta D, Marano L, Fiorentini C, Di Donato S (2003) Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology 60: 1676-9. doi: 10.1212/01.wnl.0000055872.50364.fc
Pousset F, Legrand L, Monin ML, Ewenczyk C, Charles P, Komajda M, Brice A, Pandolfo M, Isnard R, Tezenas du Montcel S, Durr A (2015) A 22-Year Follow-up Study of Long-term Cardiac Outcome and Predictors of Survival in Friedreich Ataxia. JAMA Neurol 72: 1334-41. doi: 10.1001/jamaneurol.2015.1855
Reetz K, Dogan I, Hohenfeld C, Didszun C, Giunti P, Mariotti C, Durr A, Boesch S, Klopstock T, Rodriguez de Rivera Garrido FJ, Schols L, Giordano I, Burk K, Pandolfo M, Schulz JB, Group ES (2018) Nonataxia symptoms in Friedreich Ataxia: Report from the Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology 91: e917-e930. doi: 10.1212/WNL.0000000000006121
Ribai P, Pousset F, Tanguy ML, Rivaud-Pechoux S, Le Ber I, Gasparini F, Charles P, Beraud AS, Schmitt M, Koenig M, Mallet A, Brice A, Durr A (2007) Neurological, cardiological, and oculomotor progression in 104 patients with Friedreich ataxia during long-term follow-up. Arch Neurol 64: 558-64. doi: 10.1001/archneur.64.4.558
Spieker S, Schulz JB, Petersen D, Fetter M, Klockgether T, Dichgans J (1995) Fixation instability and oculomotor abnormalities in Friedreich’s ataxia. J Neurol 242: 517-21. doi: 10.1007/bf00867423
Velasco-Sanchez D, Aracil A, Montero R, Mas A, Jimenez L, O’Callaghan M, Tondo M, Capdevila A, Blanch J, Artuch R, Pineda M (2011) Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 10: 1-8. doi: 10.1007/s12311-010-0212-7
Wallace SE, Bird TD (2018) Molecular genetic testing for hereditary ataxia: What every neurologist should know. Neurol Clin Pract 8: 27-32. doi: 10.1212/CPJ.0000000000000421
Zeigelboim BS, Mesti JC, Fonseca VR, Faryniuk JH, Marques JM, Cardoso RC, Teive HA (2017) Otoneurological Abnormalities in Patients with Friedreich’s Ataxia. Int Arch Otorhinolaryngol 21: 79-85. doi: 10.1055/s-0036-1572529
![]()