By Marcello Cherchi, MD PhD
For patients
Pendred syndrome is a genetic condition that causes hearing loss in both ears either from birth or early childhood, and often causes a goiter. The hearing loss may be identified on a newborn screening test. Imaging (an MRI of the internal auditory canals, or a CT of the temporal bone) shows inner ear malformations. Genetic testing can confirm the diagnosis. The hearing loss is often treated with cochlear implantation. The goiter may be treated with thyroid hormone supplementation. If a diagnosis of Pendred syndrome is suspected or confirmed, then it is appropriate to consult with an otolaryngologist and genetic counselor.
For clinicians
Overview
Pendred syndrome (OMIM 274600) is a genetic condition transmitted in an autosomal recessive pattern resulting from mutations in the pendrin protein encoded by part of SLC26A4 (OMIM 605646) on chromosome 7q22.3. All patients present with bilateral hearing loss either from birth (usually detected on newborn screening tests of otoacoustic emissions and brainstem auditory evoked responses) or early childhood. Most patients develop goiter, and some of those are hypothyroid. A minority of patients complain of disequilibrium (but this has not been well characterized) and suffer from acid-base disturbances. Audiometry shows bilateral approximately symmetrical sensorineural hearing loss. On vestibular testing, cervical vestibular evoked myogenic potentials may show large amplitudes and low thresholds, probably compatible with a large vestibular aqueduct. Endocrinological testing may show hypothyroidism. MRI of the brain and internal auditory canals shows a large vestibular aqueduct and cystic, incompletely formed cochlea. Genetic testing (sequencing of SLC26A4) is confirmatory. Treatment of the hearing loss is usually with cochlear implantation. Treatment for disequilibrium has not been studied. Thyroid function should be monitored, and thyroid hormone supplemented where appropriate. If Pendred syndrome is suspected or confirmed, then referral to otolaryngology and genetic counseling is appropriate.
Introduction
Dr. Vaughan Pendred (1869 – 1946) was English physician. In 1896 he published a report of two sisters, aged 28 and 38 years, who were deaf and had goiters (Pendred 1896). The original report is available at https://babel.hathitrust.org/cgi/pt?id=rul.39030030026944&view=1up&seq=588 (accessed 3/2/23).

Genetics
Pendred syndrome is caused by biallelic mutations in the SLC26A4/PDS gene (OMIM 605646) on chromosome 7q22.3 which encodes pendrin, a multi-functional anion exchanger that is found in multiple organs, including the inner ear, thyroid and kidney. The normal pendrin protein, “has affinity for chloride, iodide, and bicarbonate, among other anions” (Wemeau and Kopp 2017).
Physiology
“In the inner ear, pendrin functions as a chloride/bicarbonate exchanger that is essential for maintaining the composition and the [electrical] potential of the endolymph” (Wemeau and Kopp 2017).
“In the thyroid, pendrin is expressed at the apical membrane of thyroid cells facing the follicular lumen. Functional studies have demonstrated that pendrin can mediate iodide efflux in heterologous cells. This, together with the [pathological] thyroid phenotype observed in humans (goiter, impaired iodine organification) suggests that pendrin could be involved in iodide efflux into the lumen, one of the steps required for thyroid hormone synthesis” (Wemeau and Kopp 2017).
“In the kidney, pendrin is involved in bicarbonate secretion and chloride resorption… [It] is abundantly expressed at the apical membrane of type B, and in non‑A/non‑B intercalated cells in the cortical binding duct. Functionally, it is an apical chloride/bicarbonate exchanger mediating bicarbonate secretion, and it contributes to the regulation of blood pressure by modulating renal chloride absorption” (Wemeau and Kopp 2017).
Pathophysiology
For patients in whom genetic mutation has rendered pendrin dysfunctional, deficits in inner ear function, thyroid function and renal function may develop.
In Pendred syndrome the inner ear develops abnormally, and patients usually have a large vestibular aqueduct and a cystic, incompletely formed cochlea.
Clinical history
Patients usually present with a combination of sensorineural hearing loss and goiter, sometimes with hypothyroidism. In certain conditions these patients may also develop metabolic alkalosis.
Patients always have significant sensorineural hearing loss that is bilateral (though sometimes modestly asymmetrical) and usually congenital; less commonly it develops later in childhood.
Goiter is present in 83% of cases and usually develops after the age of 10 years; of all patients, 56% remain euthyroid, while 44% become hypothyroid, and of the latter, all have goiter (Reardon, Coffey et al. 1999). In other words, most patients develop goiter, and of the patients who develop goiter, some (but not all) are hypothyroid.
As far as renal function is concerned, “at least some patients with Pendred syndrome may develop severe acid-base disturbances under conditions of an increased alkalai load” (Reardon, Coffey et al. 1999).
A minority of Pendred syndrome patients experience disequilibrium, and this has not been well-characterized.
Testing: audiologic
The hearing loss can be detected on newborn screening with otoacoustic emissions and auditory evoked brainstem responses.
Testing: vestibular
Unfortunately, most vestibular literature pertains to findings in patients with large vestibular aqueduct. There is almost no vestibular literature on patients specifically with Pendred syndrome.
West and colleagues (West, Ryberg et al. 2021) studied video head impulse testing and cervical vestibular evoked myogenic potentials in 26 patients (52 ears) with Pendred syndrome who had not yet undergone cochlear implantation.
They reported that out of 52 ears, only 4 (8%) exhibited abnormal results on video head impulse testing; 2 ears showed low gain and 2 ears showed compensatory saccades. No patients had complete loss of the vestibulo-ocular reflex.
They reported that cervical vestibular evoked myogenic potentials (cVEMPs) responses were present in 76% of ears and absent in 24%. They calculated that the mean cVEMP amplitude was 192 microvolts, and that thresholds were <90 dB nHL.
The Figure below, from West and colleagues (West, Ryberg et al. 2021), is supposed to demonstrate abnormally large responses on cervical vestibular evoked myogenic potentials.
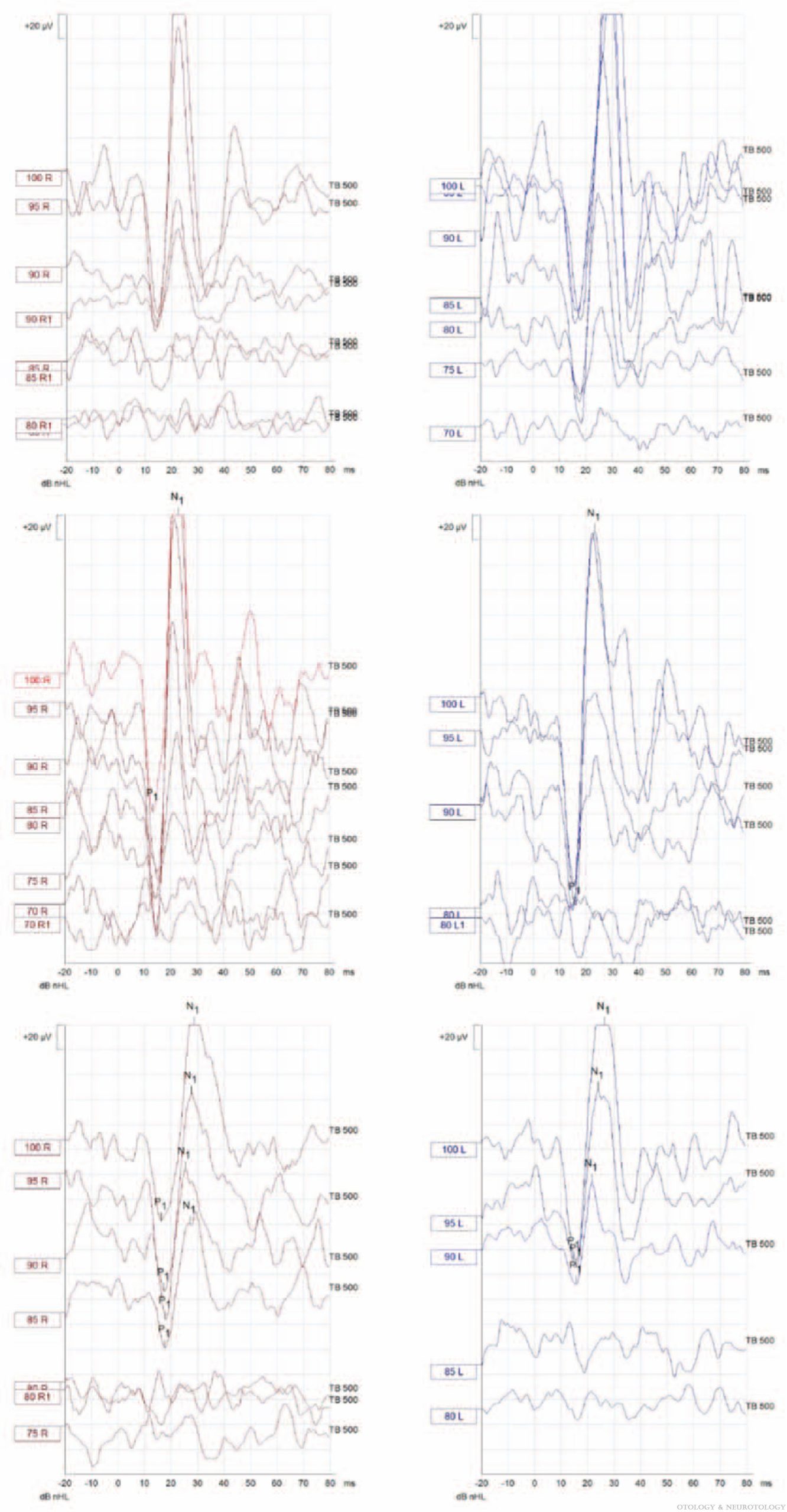
They interpreted these cVEMP findings as showing, “saccular hypersensitivity in our Pendred cohort, as the mean cVEMP thresholds were below 90 dB nHL and amplitudes were abnormally large.” Extrapolating from literature on large cochlear aqueducts, they stated that, “The cause for this hypersensitivity phenomenon has previously been suggested to be attributed to a ‘third window’ effect due to shunting of the sound-induced energy away from the cochlea.”
They further reported that, “absence of cVEMP response was associated with the presence of VHIT saccades (p=0.038),” suggesting some parallel between semicircular canal dysfunction and otolith dysfunction.
To summarize the few vestibular data on patients with Pendred syndrome: Most (92%) have normal video head impulse testing. A minority (24%) have absent cervical vestibular evoked myogenic potentials (cVEMPs); of the remainder some have abnormally large cVEMP amplitudes and low thresholds. These cVEMP findings probably reflect the presence of a large vestibular aqueduct, rather than a feature specific to Pendred syndrome.
Testing: metabolic
Checking thyroid stimulating hormone and free thyroxine can provide support for the diagnosis (Wemeau and Kopp 2017).
Testing: genetic
Definitive diagnosis can be secured by sequencing the coding region of the SLC26A4 gene (Wemeau and Kopp 2017).
Imaging
Temporal bone CT (or at least a head CT) can provide initial radiographic support for the diagnosis, though MRI of the internal auditory canals is considered the imaging modality of choice (Phelps, Coffey et al. 1998, Fugazzola, Mannavola et al. 2000).
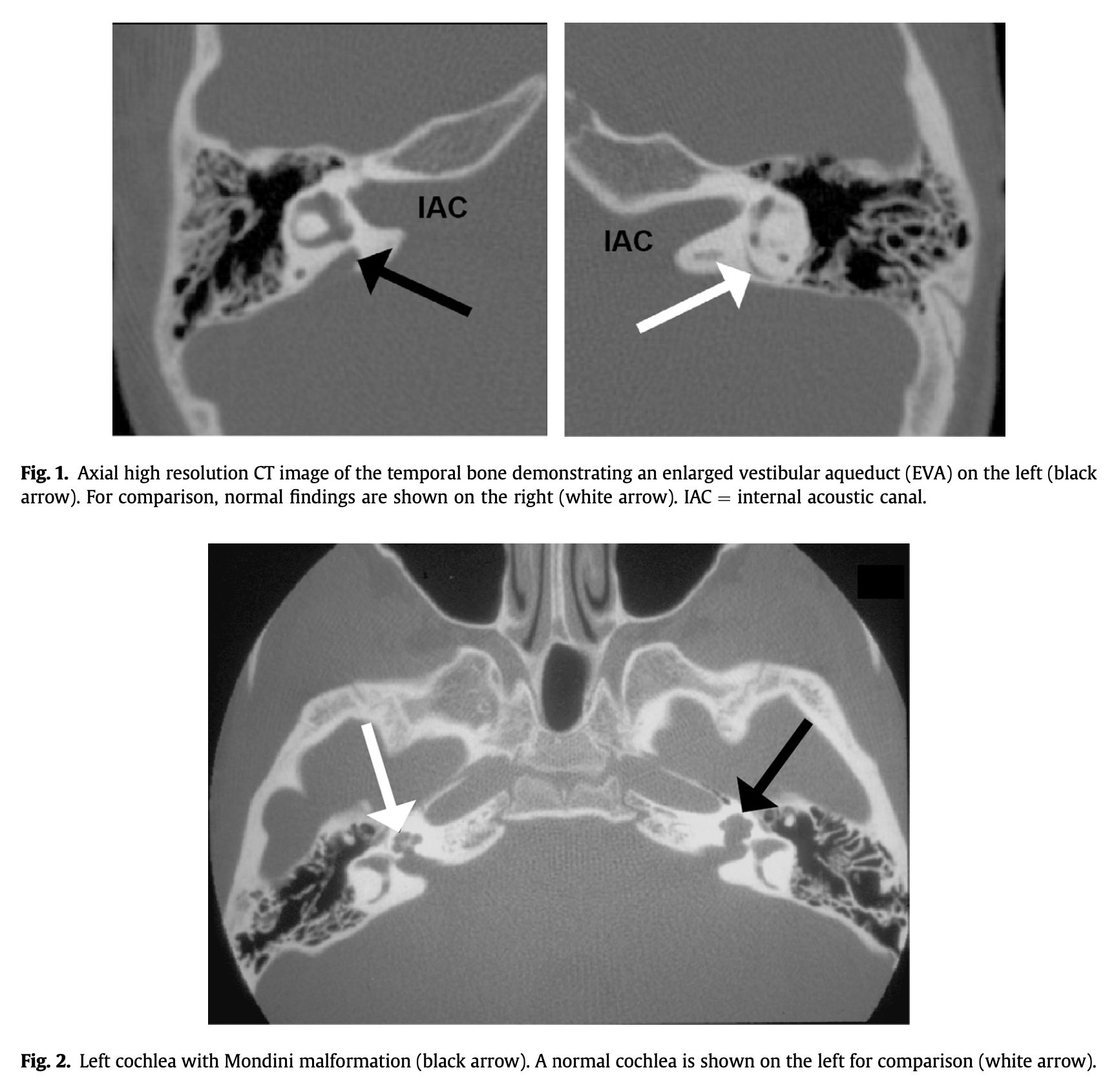
The Figure below, from Wemeau and colleagues (Wemeau and Kopp 2017), shows axial CT images demonstrating a large vestibular aqueduct on the patient’s right side (upper left panel) and an abnormal cochlea on the patient’s left (lower panel).
Treatment
Audiology consultation is appropriate. A hearing aid can be considered in patients with aidable hearing, though the majority of patients (who usually have severe to profound sensorineural hearing loss) may consider cochlear implantation.
Thyroid function should be monitored, and hypothyroidism should be treated with thyroid supplementation. Management by an endocrinologist is reasonable.
Pendred syndrome is transmitted in an autosomal recessive fashion. Referral to a genetic counselor is appropriate.
References
Pendred V (1896) Deaf-mutism and goitre. The Lancet 148: 532. doi: 10.1016/S0140-6736(01)74403-0
Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, Kendall-Taylor P, Graham JM, Cadge BC, Stephens SG, Pembrey ME, Reardon W (1998) Radiological malformations of the ear in Pendred syndrome. Clin Radiol 53: 268-73. doi: 10.1016/s0009-9260(98)80125-6
Reardon W, Coffey R, Chowdhury T, Grossman A, Jan H, Britton K, Kendall-Taylor P, Trembath R (1999) Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J Med Genet 36: 595-8.
Wemeau JL, Kopp P (2017) Pendred syndrome. Best Pract Res Clin Endocrinol Metab 31: 213-224. doi: 10.1016/j.beem.2017.04.011
West NC, Ryberg AC, Caye-Thomasen P (2021) Vestibular Function in Pendred Syndrome: Intact High Frequency VOR and Saccular Hypersensitivity. Otol Neurotol 42: e1327-e1332. doi: 10.1097/MAO.0000000000003270
![]()