By Marcello Cherchi, MD PhD
For patients
Wolfram syndrome (WS) is an inherited neurodegenerative disorder that begins in infancy or childhood with diabetes and visual loss. Some patients later develop additional problems such as hearing loss, urinary incontinence, mood disorders and other symptoms. If your doctor suspects Wolfram syndrome, then they may check several tests of vision, hearing and balance, as well as several blood tests and imaging, followed by referral to a medical genetics counselor regarding genetic testing. Hearing aids may help with the hearing loss.
For clinicians
Overview
In 1938 Dr. Don J. Wolfram (1910 – 1991) and H. Wagener published cases of four siblings with diabetes and optic atrophy (Wolfram and Wagener 1938). Subsequent descriptions added other features to this syndrome, such that it came to be known by the acronym DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy and sensorineural deafness), though much literature continues to use the eponymous designation of Wolfram syndrome (WS).
Introduction
Wolfram syndrome (WS), also known as DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy and sensorineural deafness) presents in infancy or childhood with insulin-dependent diabetes mellitus and optic atrophy. Some patients additionally develop diabetes insipidus (due to dysfunction of the supraoptic nucleus) and sensorineural hearing loss. This autosomal recessive neurodegenerative disorder with incomplete penetrance results from mutations in genes encoding either for wolframin (WS type 1) or for CDGSH iron sulfur domain protein (WS type 2). Its prevalence has been estimated at 1 in 770,000. The visual deficits are due to optic atrophy, but sometimes worsened by the presence of pigmentary retinopathy or diabetic retinopathy, and can manifest with central scotomata and constriction of peripheral vision. Visual evoked responses are usually reduced, and electroretinography amplitudes may also be reduced. Audiometry usually shows sensorineural hearing loss that is bilateral, symmetrical, and typically affects the high (greater than low) frequencies), sometimes corroborated on otoacoustic emissions (OAE). Pathologic nystagmus is reported in 8% – 75% of patients; the nystagmus is usually compatible with cerebellar dysfunction (gaze-evoked nystagmus; spontaneous up beat nystagmus), less commonly with visual loss (see-saw nystagmus). Instrumented vestibular testing may identify vestibular weakness. MRI shows prominent atrophy of the brainstem, cerebellum and anterior visual pathways (optic nerves and optic chiasm), milder cortical and subcortical atrophy with ventricular dilation. If a patient experiences disequilibrium it is likely multifactorial and due to visual, cerebellar and sometimes vestibular deficits. It is reasonable to offer amplification for the hearing loss; cochlear implantation is sometimes offered.
Epidemiology
The prevalence of WS has been estimated at 1 in 770,000 (Boutzios et al. 2011).
Genetics
WS is inherited in an autosomal recessive fashion with incomplete penetrance.
Wolfram syndrome type 1 (OMIM 222300) (WS1) is caused by a mutation in the gene encoding for wolframin (OMIM 606201) on chromosome 4p16.1. Wolframin is a transmembrane glycoprotein expressed in the endoplasmic reticulum of cells in the brain, pancreas and heart (Hofmann et al. 2003). A variety of mutations in the WS1 gene have been identified (Kobayashi et al. 2018).
Wolfram syndrome type 2 (OMIM 604928) (WS2) is caused by a mutation in the gene encoding for CISD2 (OMIM 611507). The CDGSH iron sulfur domain protein 2 (CISD2) is expressed in the endoplasmic reticulum and mitochondrial membranes (Amr et al. 2007). Other mutations in WS2 have also been reported (Amr et al. 2007).
Pathophysiological mechanism of disease
The diabetes insipidus (also known as arginine vasopressin deficiency) may result from the loss of neurons in the supraoptic nucleus that would otherwise secrete vasopressin. Gabreels and colleagues (Gabreels et al. 1998) additionally report “a defect in VP [vasopressin] precursor processing.”
The mechanism of nystagmus found in some WS patients is unknown. Al-Till and colleagues (Al-Till et al. 2002) speculate that, “Nystagmus… could be related to the underlying cerebellar degeneration.”
Clinical presentation
Affected patients usually develop insulin-dependent diabetes mellitus and optic atrophy in early childhood, less commonly as teenagers and young adults.
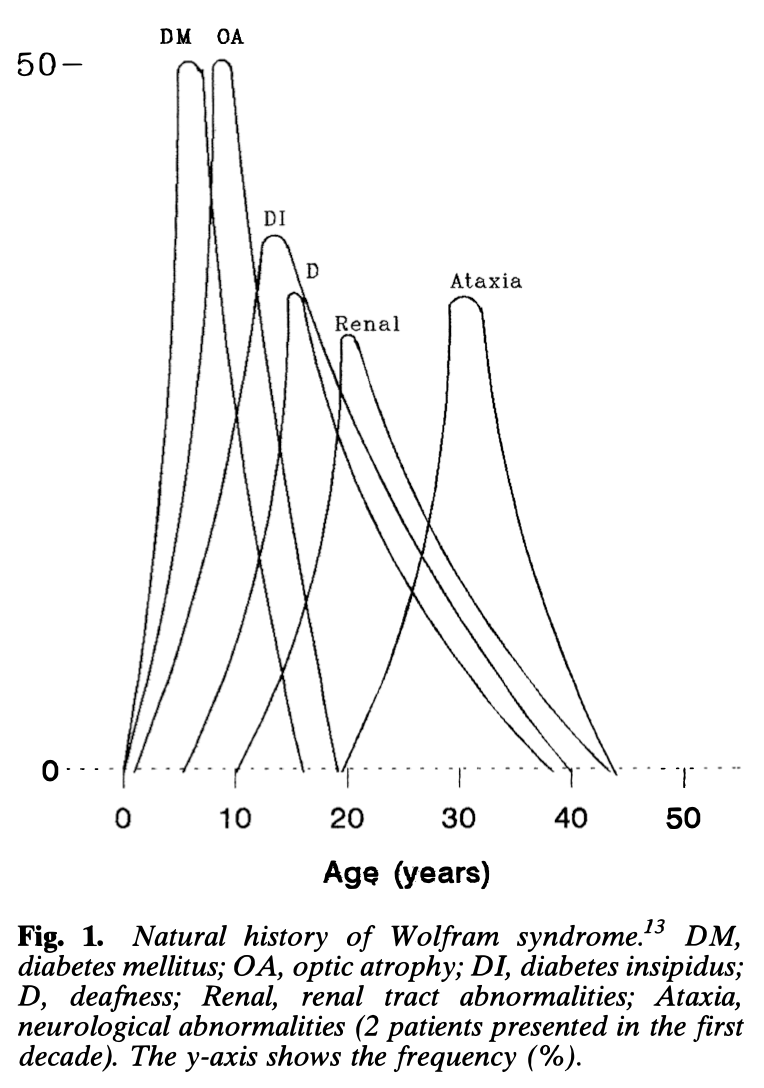
Diabetes mellitus and optic atrophy occur in all patients. The full tetrad of features occurs in 13% of patients (Barrett et al. 1997).
Barrett and colleagues (Barrett et al. 1997) report that deafness occurred in 62% (28/45) of patients, with a median onset of 16 years (range 5 – 39 years). Pennings and colleagues (Pennings et al. 2004) report that the hearing loss in WS1 is worse in females.
In the WS case series of 45 patients described by Barrett and colleagues (Barrett et al. 1997), the age of onset of each feature is displayed in the Figure below.
Additional clinical features may include genitourinary problems (neurogenic bladder, urinary incontinence, urinary tract infections, hypogonadism with decreased libido), hyposmia, psychiatric disorders (depression, anxiety, panic attacks), dysphagia, ataxia and others (Urano 2016).
The “ataxia” reported with WS is usually ascribed to cerebellar deficits (since MRI usually shows prominent cerebellar atrophy). However, if this “ataxia” is more broadly experienced as disequilibrium, then there may be more than one component. There are probably multiple mechanisms by which patients with Wolfram syndrome may experience disequilibrium, including:
- Visual loss (see below).
- Nystagmus (see below).
- Cerebellar dysfunction (see below).
- Complications from diabetes (such as diabetic peripheral neuropathy).
Ocular motor examination
Most descriptions of WS make no comment about nystagmus, and the scarcity of observations may account for the variability in its reported frequency, ranging from 8% (Al-Till et al. 2002) to 28% (Marshall et al. 2013) to 75% (Gunn et al. 1976).
Most of the literature on WS does not describe the associated nystagmus well. For example:
- Gunn and colleagues (Gunn et al. 1976) reported nystagmus in 75% (3/4) patients, but did not provide further details.
- Marshall and colleagues (Marshall et al. 2013) studied a series of 18 WS1 patients and reported “abnormal… nystagmus” in 28% (5/18), without further details.
Some studies report “horizontal nystagmus” or “end-gaze nystagmus,” such as:
- Al-Till and colleagues say that one patient in their series had “horizontal jerky nystagmus” (Al-Till et al. 2002).
- Barrett and colleagues (Barrett et al. 1997) state that out of 11 patients with absent pupillary light reflexes, 9 had “horizontal nystagmus,” adding that, “these were older patients with other cerebellar signs including ataxia and dysarthria.”
- Kabanovski and colleagues studied 5 WS patients and reported “end-gaze nystagmus” (Kabanovski et al. 2022).
Other studies additionally report vertical or torsional nystagmus:
- Grosse Aldenhovel and colleagues (Grosse Aldenhovel et al. 1991) reported one patient who, “had vertical nystagmus in the primary position of the eyes and on up-gaze as well as horizontal nystagmus to both sides.”
- Hoekel and colleagues (Hoekel et al. 2014) studied a series of 18 WS patients and reported nystagmus in 39% (7/18). They further commented, “Nystagmus was observed in 7 subjects (39%), 6 of whom had horizontal conjugate jerk nystagmus when gaze was directed more than 15˚ from primary gaze (gaze-evoked nystagmus). One patient (6%) had rotary/torsional/see-saw nystagmus.”
- Rando and colleagues (Rando et al. 1992) reported on two WS patients, of whom one had “clockwise rotatory nystagmus” and the other had “gaze-evoked horizontal nystagmus.”
- Megighian and Savastano (Megighian and Savastano 2004) reported a case of WS in which, “Two types of nystagmus are present: one is a horizontal jerky nystagmus towards the left with a rotary component and the other one is an upbeat vertical nystagmus.” They also reported that horizontal optokinetic nystagmus was “bilaterally altered” (without further details) and vertical optokinetic responses were normal.
Most of these ocular motor findings (gaze-evoked horizontal nystagmus, spontaneous up beat nystagmus) are compatible with cerebellar dysfunction. The finding of see-saw nystagmus may reflect the visual loss associated with optic nerve and optic chiasm atrophy.
Testing: auditory
Audiometry shows approximately symmetrical sensorineural hearing loss predominantly in the high frequencies. The Figure below from Higashi (Higashi 1991) shows a composite audiogram of multiple patients.
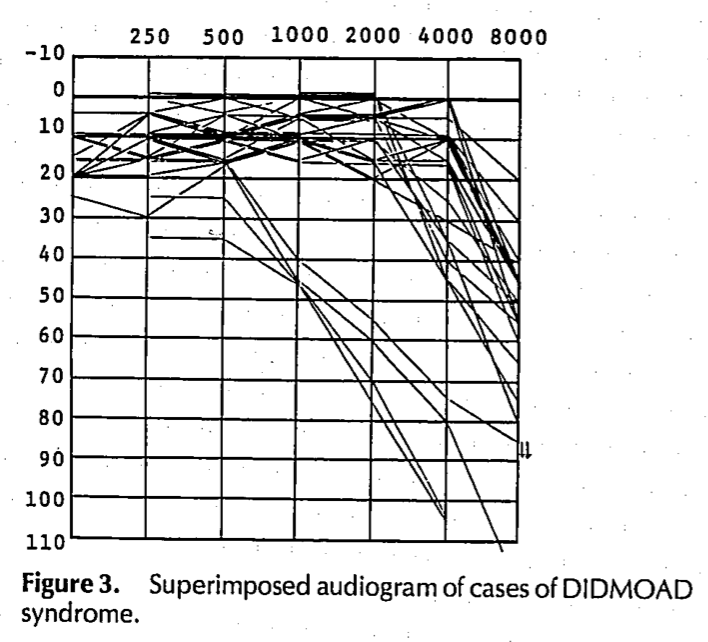
Less commonly, low frequencies are affected greater than high frequencies (Jung et al. 2024).
Fujikawa and colleagues reported on two WS patients and found that “DPOAE [distortion product otoacoustic emissions] amplitudes were decreased to the noise level only at the mid-frequencies… which corresponded to the audiometric configurations” in both ears of each patient (Fujikawa et al. 2010).
Pennings and colleagues (Pennings et al. 2004) reported on a series of 11 WS1 patients. Of these, two underwent auditory brainstem evoked responses (ABR), and the results were reported as normal.
Testing: vestibular
Fujikawa and colleagues reported on two WS patients and found SP/AP ratios were increased (Fujikawa et al. 2010) on electrocochleography (ECoG).
Fujikawa and colleagues reported that in one WS patient tested, “a normal caloric response was observed for both ears”(Fujikawa et al. 2010), and that this was tested with air calorics.
Karzon and Hullar studied a series of 11 WS patients (Karzon and Hullar 2013), of whom three underwent rotatory chair testing (RCT). They reported that:
- On slow harmonic acceleration:
- One patient had “abnormally low gains” and “low-frequency phase lead was pathologically elevated,” which suggests vestibular weakness.
- One patient had “borderline low gains across frequencies tested.”
- On step velocity testing:
- All three patients “had borderline low gains in at least one direction.”
- One patient “had a markedly decreased time constant in both directions.”
In short, the RCT findings reported by Karzon and Hullar suggest some degree of vestibular weakness, but they added, “Our finding that profound bilateral vestibular loss was not common is typical of patients with WS” (Karzon and Hullar 2013). On review of other literature, Karzon and Hullar found only one report (Barjon et al. 1964) of a patient whose vestibular loss was more pronounced than the sensorineural hearing loss.
Pennings and colleagues (Pennings et al. 2004) reported on a series of 11 WS1 patients, of whom six “underwent vestibular examination with a rotatory chair and electronystagmography.” Of those six patients, “One individual had a hyperactive vestibulo-ocular reflex with asymmetry. One individual… had vestibular areflexia and an enhanced cervico-ocular reflex.”
Testing: other
WS patients generally have one or more factors impairing vision.
Al-Till and colleagues (Al-Till et al. 2002) report optic atrophy in 93% of eyes, pigmentary retinopathy in 30% of eyes and diabetic retinopathy in 20% of eyes.
On visual field testing, the initial visual deficits from optic atrophy include central scotomata with peripheral constriction (Barrett et al. 1997).
Barrett and colleagues (Barrett et al. 1997) reported that electroretinography showed reduced amplitude in 50% (3 out of 6) patients.
Barrett and colleagues (Barrett et al. 1997) reported that visual evoked responses were abnormal in 100% (10 out of 10 patients, showing reduced amplitude to flash and pattern stimulation.
Imaging
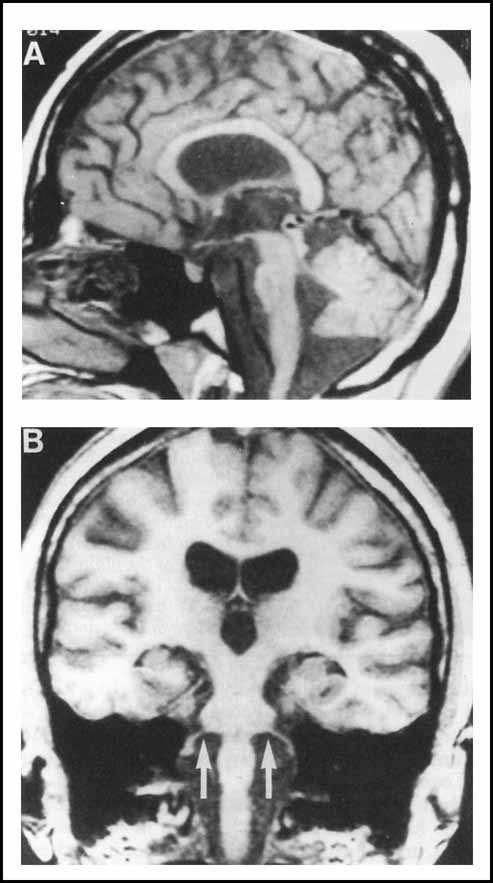
Barrett and colleagues (Barrett et al. 1997) reported that out of 11 patients who underwent brain MRI, 8 (73%) showed abnormalities, and “the commonest finding was a generalized atrophy, particularly of the brainstem and cerebellum.”
Rando and colleagues (Rando et al. 1992) reported imaging findings in two WS patients as follows:
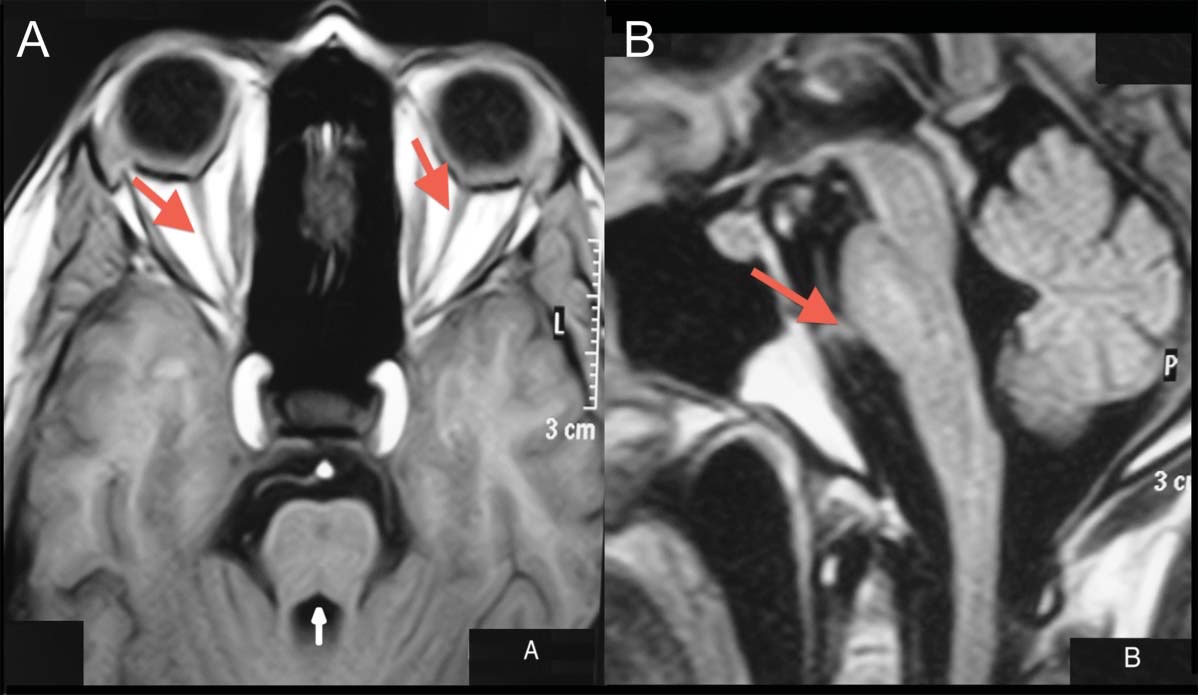
- “MRI revealed moderate atrophy of the brainstem and cerebellum… There was mild, diffuse atrophy of the cerebral hemispheres, and the entire ventricular system was dilated. With narrow cuts through the midline, the anterior visual pathway appeared shrunken, thinning of the hypothalamic region was evident, and the posterior lobe of the pituitary could not be identified.”
- “MRI… revealed atrophy of the brainstem and the cerebellum. There was mild, diffuse cortical atrophy and the ventricular system was moderately dilated. The optic nerves and chiasm, the hypothalamus, and the pituitary gland appeared atrophic.”
To summarize, brain MRI shows prominent atrophy of the brainstem, cerebellum and anterior visual pathway (optic nerves and optic chiasm), and milder cortical and subcortical atrophy with ventricular dilation. Representative images are shown in the Figures below.
Histopathology
Barrett and colleagues (Barrett et al. 1997) reported that autopsy findings in one patient “revealed atrophy of the optic nerves, chiasm, cerebellum and brainstem.”
Differential diagnosis
Referral to a medical genetics counselor regarding genetic testing for WS is appropriate.
The differential diagnosis of Wolfram syndrome includes several mitochondrial disorders, autosomal dominant optic nerve atrophy, Friedreich ataxia, Bardet-Biedl syndrome and Alstrom syndrome (Urano 2016).
Treatment
There is no curative or arrestive treatment. Amplification may help with the hearing loss. Cochlear implantation is sometimes offered (Alías et al. 2022; Fehrmann et al. 2024; Lim et al. 2023; Marfatia et al. 2024).
Prognosis
The prognosis is poor.
References
Al-Till M, Jarrah NS, Ajlouni KM (2002) Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur J Ophthalmol 12: 84-8. doi: 10.1177/112067210201200202
Alías L, López de Heredia M, Luna S, Clivillé N, González-Quereda L, Gallano P, de Juan J, Pujol A, Diez S, Boronat S, Orús C, Lasa A, Venegas MDP (2022) Case report: De novo pathogenic variant in WFS1 causes Wolfram-like syndrome debuting with congenital bilateral deafness. Front Genet 13: 998898. doi: 10.3389/fgene.2022.998898
Amr S, Heisey C, Zhang M, Xia XJ, Shows KH, Ajlouni K, Pandya A, Satin LS, El-Shanti H, Shiang R (2007) A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet 81: 673-83. doi: 10.1086/520961
Barjon P, Lestradet H, Labauge R (1964) [Primary Optic Atrophy and Neurogenic Deafness in Juvenile Diabetes. (Apropos of 3 Cases)]. Presse Med (1893) 72: 983-6.
Barrett TG, Bundey SE, Fielder AR, Good PA (1997) Optic atrophy in Wolfram (DIDMOAD) syndrome. Eye (Lond) 11 ( Pt 6): 882-8. doi: 10.1038/eye.1997.226
Boutzios G, Livadas S, Marinakis E, Opie N, Economou F, Diamanti-Kandarakis E (2011) Endocrine and metabolic aspects of the Wolfram syndrome. Endocrine 40: 10-3. doi: 10.1007/s12020-011-9505-y
Fehrmann MLA, Lanting CP, Haer-Wigman L, Mylanus EAM, Huinck WJ, Pennings RJE (2024) Good cochlear implantation outcomes in subjects with mono-allelic WFS1-associated sensorineural hearing loss – a case series. Int J Audiol: 1-9. doi: 10.1080/14992027.2024.2411579
Fujikawa T, Noguchi Y, Ito T, Takahashi M, Kitamura K (2010) Additional heterozygous 2507A>C mutation of WFS1 in progressive hearing loss at lower frequencies. Laryngoscope 120: 166-71. doi: 10.1002/lary.20691
Gabreels BA, Swaab DF, de Kleijn DP, Dean A, Seidah NG, Van de Loo JW, Van de Ven WJ, Martens GJ, Van Leeuwen FW (1998) The vasopressin precursor is not processed in the hypothalamus of Wolfram syndrome patients with diabetes insipidus: evidence for the involvement of PC2 and 7B2. J Clin Endocrinol Metab 83: 4026-33. doi: 10.1210/jcem.83.11.5158
Grosse Aldenhovel HB, Gallenkamp U, Sulemana CA (1991) Juvenile onset diabetes mellitus, central diabetes insipidus and optic atrophy (Wolfram syndrome)–neurological findings and prognostic implications. Neuropediatrics 22: 103-6. doi: 10.1055/s-2008-1071426
Gunn T, Bortolussi R, Little JM, Andermann F, Fraser FC, Belmonte MM (1976) Juvenile diabetes mellitus, optic atrophy, sensory nerve deafness, and diabetes insipidus–a syndrome. J Pediatr 89: 565-70. doi: 10.1016/s0022-3476(76)80387-3
Higashi K (1991) Otologic findings of DIDMOAD syndrome. Am J Otol 12: 57-60.
Hoekel J, Chisholm SA, Al-Lozi A, Hershey T, Tychsen L, Washington University Wolfram Study G (2014) Ophthalmologic correlates of disease severity in children and adolescents with Wolfram syndrome. J AAPOS 18: 461-465 e1. doi: 10.1016/j.jaapos.2014.07.162
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF (2003) Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 12: 2003-12. doi: 10.1093/hmg/ddg214
Jung J, Jang SH, Won D, Gee HY, Choi JY, Jung J (2024) Clinical Characteristics and Audiological Profiles of Patients with Pathogenic Variants of WFS1. J Clin Med 13. doi: 10.3390/jcm13164851
Kabanovski A, Donaldson L, Margolin E (2022) Neuro-ophthalmological manifestations of Wolfram syndrome: Case series and review of the literature. J Neurol Sci 437: 120267. doi: 10.1016/j.jns.2022.120267
Karzon RK, Hullar TE (2013) Audiologic and vestibular findings in Wolfram syndrome. Ear Hear 34: 809-12. doi: 10.1097/AUD.0b013e3182944db7
Kobayashi M, Miyagawa M, Nishio SY, Moteki H, Fujikawa T, Ohyama K, Sakaguchi H, Miyanohara I, Sugaya A, Naito Y, Morita SY, Kanda Y, Takahashi M, Ishikawa K, Nagano Y, Tono T, Oshikawa C, Kihara C, Takahashi H, Noguchi Y, Usami SI (2018) WFS1 mutation screening in a large series of Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis. PLoS One 13: e0193359. doi: 10.1371/journal.pone.0193359
Lim HD, Lee SM, Yun YJ, Lee DH, Lee JH, Oh SH, Lee SY (2023) WFS1 autosomal dominant variants linked with hearing loss: update on structural analysis and cochlear implant outcome. BMC Med Genomics 16: 79. doi: 10.1186/s12920-023-01506-x
Marfatia H, Rattan A, Jain A (2024) Cochlear implant in Wolfram syndrome: A case report. Cochlear Implants Int 25: 487-491. doi: 10.1080/14670100.2024.2442826
Marshall BA, Permutt MA, Paciorkowski AR, Hoekel J, Karzon R, Wasson J, Viehover A, White NH, Shimony JS, Manwaring L, Austin P, Hullar TE, Hershey T (2013) Phenotypic characteristics of early Wolfram syndrome. Orphanet J Rare Dis 8: 64. doi: 10.1186/1750-1172-8-64
Megighian D, Savastano M (2004) Wolfram syndrome. Int J Pediatr Otorhinolaryngol 68: 243-7. doi: 10.1016/j.ijporl.2003.10.012
Pennings RJ, Huygen PL, van den Ouweland JM, Cryns K, Dikkeschei LD, Van Camp G, Cremers CW (2004) Sex-related hearing impairment in Wolfram syndrome patients identified by inactivating WFS1 mutations. Audiol Neurootol 9: 51-62. doi: 10.1159/000074187
Rando TA, Horton JC, Layzer RB (1992) Wolfram syndrome: evidence of a diffuse neurodegenerative disease by magnetic resonance imaging. Neurology 42: 1220-4. doi: 10.1212/wnl.42.6.1220
Urano F (2016) Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr Diab Rep 16: 6. doi: 10.1007/s11892-015-0702-6
Vale TC, Perpetuo FO (2013) Teaching NeuroImages: a neuroendocrine rarity: Wolfram syndrome. Neurology 81: e153. doi: 10.1212/01.wnl.0000435559.06072.d6
Wolfram DJ, Wagener H (1938) Diabetes mellitus and simple optic atrophy among siblings: report on four cases. Mayo Clinic Proceedings 13: 715-718.
![]()