By Marcello Cherchi, MD PhD
For patients
Multiple system atrophy (MSA) is a degenerative process affecting the brain and other organ systems. There are several subtypes that can cause various combinations of findings, including unsteadiness, clumsiness, trouble pronouncing words and fainting. MSA can be difficult to distinguish from several other diseases, but this distinction may be clarified through various tests such as a brain MRI or other special brain scan, testing of eye movements, tests of cardiac function, and others, though sometimes even after doing such tests the diagnosis remains unclear. There is no treatment that stops or reverses the disease. From the time of symptom onset, most patients die within about 10 years.
For clinicians
Overview
Multiple system atrophy (MSA) is a sporadically occurring progressive neurodegenerative alpha synucleinopathy that causes various combinations of parkinsonism, ataxia and autonomic dysfunction. Several subtypes have been reported according to which feature is most prominent. The disease is rare (0.6 to 0.7 cases per 100,000 person years), with a peak age of onset in the 6th decade. Common presenting symptoms include falls, dysphagia and REM sleep behavior disorder. Depending on the subtype, physical examination may show findings compatible with parkinsonism or ataxia. Face-to-face ocular motor examination may reveal spontaneous nystagmus, gaze-evoked nystagmus and choppy smooth pursuit. Instrumented ocular motor testing may additionally show square wave jerks, fixation abnormalities, saccadic dysmetria and reduced vestibulo-ocular reflex. Vestibular evoked myogenic potentials may be reduced or absent. Autonomic testing may reveal orthostatic hypotension and abnormalities on thermoregulatory sweat testing (less commonly on quantitative sudomotor axon reflex test). Brain MRI may show the so-called “hot cross buns” sign, or “putaminal slit” sign. FDG‑PET may show hypometabolism in the putamina. The differential diagnosis is the other parkinsonian disorders. There is no curative or arrestive treatment, so management is symptomatic and often involves multiple subspecialties (neurology, psychiatry, cardiology, gastroenterology, urology, physical and occupational therapy). Prognosis is poor, with a mean survival from symptom onset of 9.8 years.
Introduction
Multiple system atrophy (MSA) is a sporadically occurring alpha synucleinopathy that manifests as a relentlessly progressive neurodegenerative disorder, and whose subtypes exhibit varying degrees of parkinsonism, ataxia and autonomic dysfunction (Fanciulli and Wenning 2015, Fanciulli, Stankovic et al. 2019). Three main forms have been described.
- Dr. Joseph Jules Dejerine and Dr. Andre Thomas reported on a patient with what they termed olivopontocerebellar atrophy (OPCA) (Dejerine and Thomas 1900).
- Dr. Raymond Delacy Adams (1911 – 2008), Dr. Ludo Van Bogaert (1897 – 1989) and Dr. Henri Marie Hector Vander Eecken (born 1920), described what eventually became known as striatonigral degeneration (SND) (Adams, Vanbogaert et al. 1964).
- Dr. George Milton Shy and Dr. Glenn Albert Drager reported a series of patients with autonomic failure (Shy and Drager 1960). For a time this was known as Shy-Drager syndrome (SDS).
Dr. J.G. Graham and Dr. D.R. Oppenheimer first used the term multiple system atrophy (MSA) as an umbrella term for these various syndromes (Graham and Oppenheimer 1969).
Since then a number of other “atypical variants” of MSA have been described (Jellinger 2022), including:
- MSAs with greater than expected cognitive impairment.
- MSA with Lewy body disease. Since MSA and Lewy body disease are both alpha synucleinopathies, this clinical entity seems more likely to be one in which the neuropathology from one disease extends beyond its usual neuroanatomical area, provoking additional symptoms suggestive of the other disease.
- MSA with cognitive impairment/dementia. This sounds similar to the “MSA with Lewy body disease” mentioned earlier.
- MSA with severe hippocampal atrophy.
- MSAs with additional motor symptoms.
- MSA with dystonia.
- MSA with spinal myoclonus.
- MSA with tau pathology. We find it difficult to understand how this would be classified as an “atypical MSA” since MSA is an alpha synucleinopathy rather than a tauopathy. We think this entity is more likely to be an “atypical tauopathy” whose pathology unusually happens to be in the neuroanatomical distribution that causes an MSA-like clinical picture.
The different presentations and deficits in MSA are likely due to variability in the distribution and degree of neuropathological involvement (Fanciulli and Wenning 2015). Multiple organ systems can be affected in MSA, as summarized in the Figure below from Fanciulli and colleagues (2015).
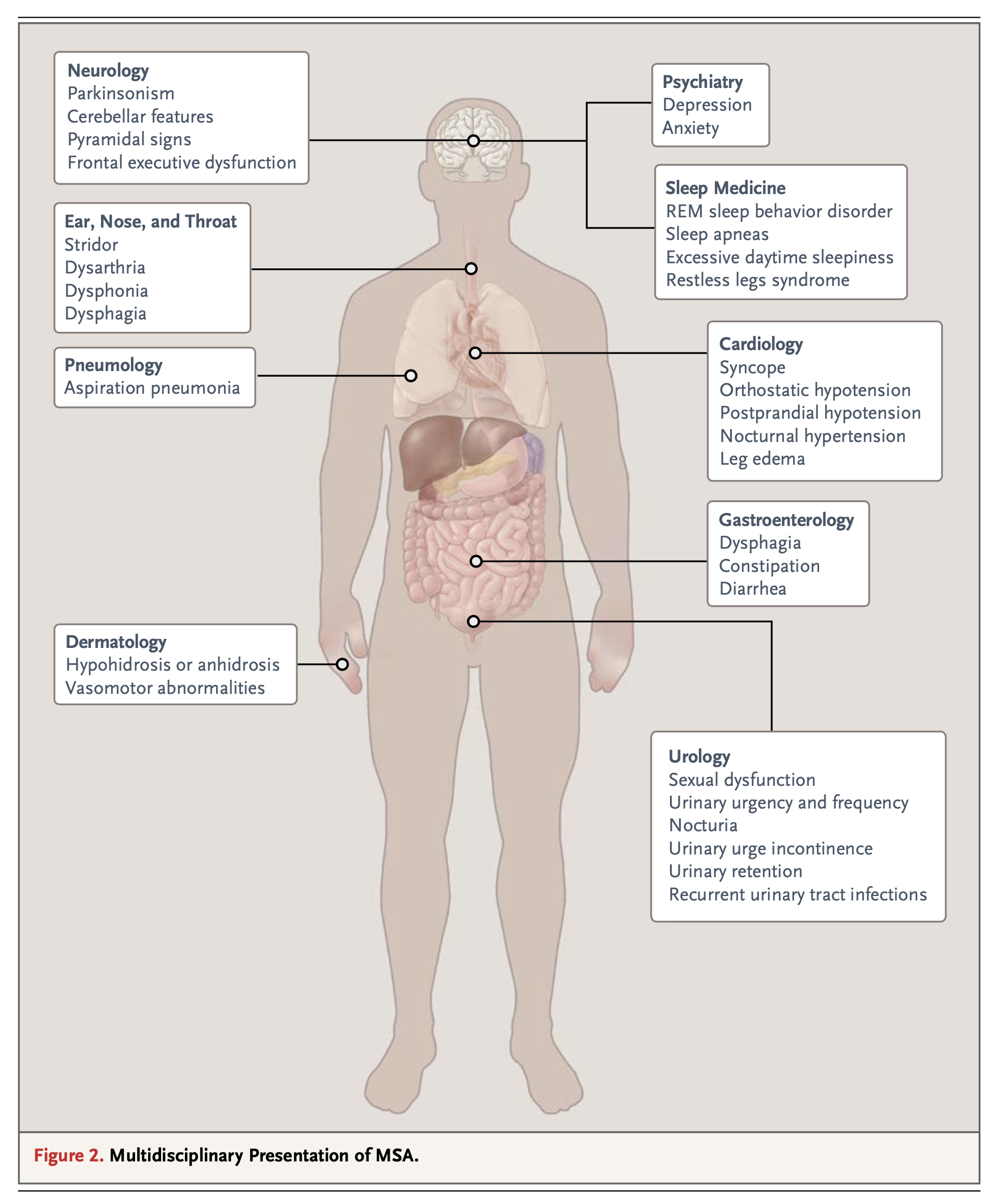
Epidemiology
The mean incidence of MSA is 0.6 to 0.7 cases (range 0.1 – 2.4 cases) per 100,000 person-years. The point prevalence is 3.4 – 4.9 cases per 100,000 people, increasing to 7.8 per 1000,000 people older than 40 years (Fanciulli and Wenning 2015).
The peak age of onset of MSA is in the 6th decade, though earlier (4th – 5th decade) and later (8th decade) onsets have been reported (Fanciulli, Stankovic et al. 2019). So-called “young onset MSA” is rare; Kim and colleagues (Kim, Jeon et al. 2012) studied 455 MSA patients and reported that 4 (0.9%) had symptom onset prior to the age of 40 years. Late-onset MSA is similarly unusual; Lee and colleagues (Lee, Ando et al. 2020) studied 1425 MSA patients and reported that 39 (2.7%) developed first symptoms at or after 76.8 years.
Most cases of MSA appear to be sporadic. Several pedigrees have been reported in which MSA appears to occur in an autosomal dominant or autosomal recessive fashion (Fanciulli, Stankovic et al. 2019, Jellinger 2022).
Pathophysiological mechanism of disease
The underlying pathobiology of MSA remains incompletely understood (Fanciulli, Stankovic et al. 2019). The normal alpha synuclein protein appears to be involved in synaptic vesicle transport and neurotransmitter release (Monzio Compagnoni and Di Fonzo 2019). The abnormal alpha synuclein protein appears to have a predilection for oligodendrocytes (Ahmed, Asi et al. 2012, Jellinger 2018).
Clinical presentation
Synucleinopathies are often associated with REM sleep behavior disorder (RBD). MSA is no exception, and often the RBD precedes other symptoms by years. Iranzo and colleagues (Iranzo, Santamaria et al. 2005) studied 26 MSA patients and found that 54% had RBD prior to developing any waking motor symptoms.
O’Sullivan and colleagues (O’Sullivan, Massey et al. 2008) reviewed the records of 83 cases of MSA and reported symptom onset (differentiated by presence/absence of early autonomic symptoms) as follows:
|
MSA with early autonomic symptoms |
MSA without early autonomic symptoms |
|||
|
Number (percent) |
Years since initial symptom onset |
Number (percent) |
Years since initial symptom onset |
|
|
Frequent falls |
22 (55%) |
5.7 ± 2.2 |
23 (64%) |
5.4 ± 2.4 |
|
Wheelchair dependent |
23 (55%) |
6.6 ± 2.5 |
19 (56%) |
6.9 ± 1.8 |
|
Unintelligible speech |
14 (33%) |
6.7 ± 2.0 |
19 (53%) |
7.6 ± 2.0 |
|
Severe dysphagia |
13 (32%) |
7.1 ± 2.4 |
12 (33%) |
7.6 ± 2.3 |
|
Urinary catheter |
29 (71%) |
5.8 ± 2.7 |
18 (50%) |
6.9 ± 2.3 |
|
Cognitive impairment |
5 (13%) |
6.6 ± 2.5 |
5 (14%) |
6.6 ± 2.0 |
In the MSA‑A variant, early autonomic dysfunction occurs in 60% of patients, and can involve erectile dysfunction, lower urinary tract symptoms, anhidrosis and cardiovascular autonomic symptoms such as recurrent syncope (Fanciulli, Stankovic et al. 2019).
The MSA‑C and MSA‑P variants are distinguished by whether the predominant motor features are cerebellar (MSA‑C) or parkinsonian (MSA‑P) (Gilman, Wenning et al. 2008).
Physical examination
On physical examination patients have some combination of parkinsonism and ataxia. Bedside examination of orthostatics may reveal orthostatic hypotension.
Ocular motor examination
MSA patients may exhibit spontaneous nystagmus or gaze-evoked nystagmus (Fanciulli, Stankovic et al. 2019).
Anderson and colleagues (Anderson, Luxon et al. 2008) studied 30 patients with probable MSA (22 with MSA‑P and 8 with MSA‑C) by clinical examination. They reported “excessive square wave jerks” in 70% (21/30), “mild supranuclear gaze palsy” in 27% (8/30), gaze-evoked nystagmus in 40% (12/30), “positioning down beat nystagmus” in 40% (10/25), mild or moderate saccadic hypometria in 73% (22/30), “broken up” smooth pursuit in 93% (28/30) and reduced vestibulo-ocular reflex suppression in 67% (16/24). They further reported that formal videonystagmography with caloric testing revealed no additional findings.
Testing: videonystagmography (VNG)
Pinkhardt and colleagues (Pinkhardt, Kassubek et al. 2009) studied instrumented ocular motor testing in 19 MSA patients (11 with MSA‑P and 8 with MSA‑C) and compared them with 27 patients with idiopathic Parkinson’s disease (IPD) and 23 healthy controls. They reported that smooth pursuit eye movements were similar between the MSA‑P and MSA‑C patients, but there was significant overlap with the IPD and healthy controls.
Pinnock and colleagues (Pinnock, McGivern et al. 2010) studied instrumented ocular motor testing in 44 patients with various parkinsonian disorders (MSA, progressive supranuclear palsy and idiopathic Parkinson’s disease) compared to 50 age-matched healthy controls. They reported that the patients “exhibited larger and more frequent SI [saccadic intrusions] as well as greater displacement from the fixation target,” adding that, “Patients with multiple system atrophy show increased frequency of SI both with and without a visible target.”
Klotz and Klockgether (Klotz and Klockgether 2005) reported a case of an MSA patient with very prominent macro square wave jerks.
Zhou and colleagues (Zhou, Sun et al. 2022) studied instrumented ocular motor testing in 45 patients with MSA (24 with MSA‑P and 21 with MSA‑C) and 40 age-matched healthy controls. They reported that overall 93% of MSA patients exhibited ocular motor abnormalities, broken down as follows: 38% had abnormalities on fixation and gaze holding; 51% had abnormalities without fixation; 73% had abnormalities on saccades; 71% had abnormalities on smooth pursuit; and 38% had abnormalities on optokinetic testing.
Zhou and colleagues (Zhou, Wang et al. 2021) studied instrumented ocular motor testing in 33 MSA patients (18 with MSA‑P and 15 with MSA‑C), 96 patients with idiopathic Parkinson’s disease, and 40 healthy controls. They reported that 64% of MSA patients exhibited saccadic dysmetria (both hypometria and hypermetria), and that this was statistically significantly greater than the other groups.
Brooks and colleagues (Brooks, Klier et al. 2017) studied prosaccade and anti-saccade tasks in 11 MSA patients and compared them to 21 patients with idiopathic Parkinson’s disease and 20 healthy controls. They reported that MSA patients had longer prosaccade latencies than the other groups.
Venhovens and colleagues (Venhovens, Meulstee et al. 2020) studied 6 patients with MSA. They reported that 6 (100%) had saccadic abnormalities, 6 (100%) had abnormal smooth pursuit, 5 (83%) had optokinetic abnormalities, 4 (67%) had abnormalities of fixation, 2 (33%) had abnormalities on rotatory chair testing, and 1 (17%) had spontaneous nystagmus.
In the Table below we have compiled the various ocular motor abnormalities from both instrumented and face-to-face studies.
|
Anderson et al. 2008 |
Zhou et al. 2022 |
Zhou et al. 2021 |
Venhovens et al. 2020 |
Klotz et al. 2005 |
Brooks et al. 2017 |
Pinkhardt et al. 2009 |
Pinnock et al. 2010 |
|
|
Supranuclear gaze palsy |
27% |
|||||||
|
Nystagmus, gaze evoked |
40% |
|||||||
|
Nystagmus, positioning down beat |
40% |
|||||||
|
Nystagmus, spontaneous |
17% |
|||||||
|
Saccades, “abnormal” |
73% |
100% |
||||||
|
Saccadic dysmetria (hyper‑ and hypo‑) |
64% |
|||||||
|
Saccadic hypometria |
73% |
|||||||
|
Saccades, prolonged prosaccadic latencies |
√ |
|||||||
|
Smooth pursuit, “broken up” |
93% |
71% |
100% |
√ |
||||
|
Vestibulo-ocular reflex, reduced |
67% |
|||||||
|
Square wave jerks |
70% |
√ |
||||||
|
Saccadic intrusions |
√ |
|||||||
|
Fixation and gaze holding abnormal |
38% |
67% |
||||||
|
Gaze abnormal without fixation (perhaps spontaneous nystagmus?) |
51% |
|||||||
|
Optokinetic nystagmus abnormal |
38% |
83% |
||||||
|
Abnormalities on rotatory chair testing |
33% |
Table : Ocular motor abnormalities reported in multiple system atrophy
In summary, in studies that report frequency of occurrence, the ocular motor abnormalities found in ≥50% of patients with MSA include square wave jerks and other fixation abnormalities, gaze holding abnormalities, saccadic abnormalities (usually dysmetria), choppy smooth pursuit and reduced vestibulo-ocular reflex.
Testing: vestibular evoked myogenic potentials (VEMPs)
Venhovens and colleagues (Venhovens, Meulstee et al. 2020) studied 6 patients (12 ears) with MSA. On cervical vestibular evoked myogenic potentials, responses were delayed unilaterally in 2 (33%) patients and bilaterally in 1 (17%) patient; responses were absent in 1 patient (17%). On ocular vestibular evoked myogenic potentials, responses were absent unilaterally in 3 (50%) patient and bilaterally in 1 (17%) patient.
Scarpa and colleagues (Scarpa, Cassandro et al. 2020) studied 16 MSA patients with cervical vestibular evoked myogenic potentials and reported them to be absent bilaterally in 27% and unilaterally in 13% of patients.
Testing: subjective visual vertical (SVV)
Venhovens and colleagues (Venhovens, Meulstee et al. 2020) studied 6 patients with MSA and reported that 1 (17%) had an abnormal response on subjective visual vertical testing.
Testing: other
Given the pronounced autonomic dysfunction in many cases of MSA, orthostatic hypotension is often detectable at the bedside. If bedside testing does not identify this yet clinical suspicion for MSA is high, then checking a tilt table test is reasonable.
Up to 80% of patients with MSA have decreased sweating, and up to 45% have frank anhidrosis (Iodice, Lipp et al. 2012). The autonomic dysfunction is usually attributed to the disease’s preganglionic deficits (Donadio, Nolano et al. 2008). However, mounting evidence suggests that MSA involves post-ganglionic autonomic fibers as well (Provitera, Nolano et al. 2014, Doppler, Weis et al. 2015, Coon, Fealey et al. 2017). Thus, thermoregulatory sweat testing (TST) (which assesses both pre- and post-ganglionic autonomic function) is abnormal in most cases of MSA, whereas quantitative sudomotor axon reflex test (QSART) (which only assesses post-ganglionic autonomic function) is only abnormal in about 30% of cases (Palma, Norcliffe-Kaufmann et al. 2018).
Imaging
The usual diagnostic dilemma is in distinguishing MSA from other parkinsonian syndromes such as idiopathic Parkinson’s disease, progressive supranuclear palsy and corticobasal ganglionic degeneration. Radiographic findings are sometimes helpful in this regard, but imperfect (Schrag, Good et al. 2000).
Some brain MRI findings have reasonably good predictive values for a diagnosis of MSA (Fanciulli, Stankovic et al. 2019). These include:
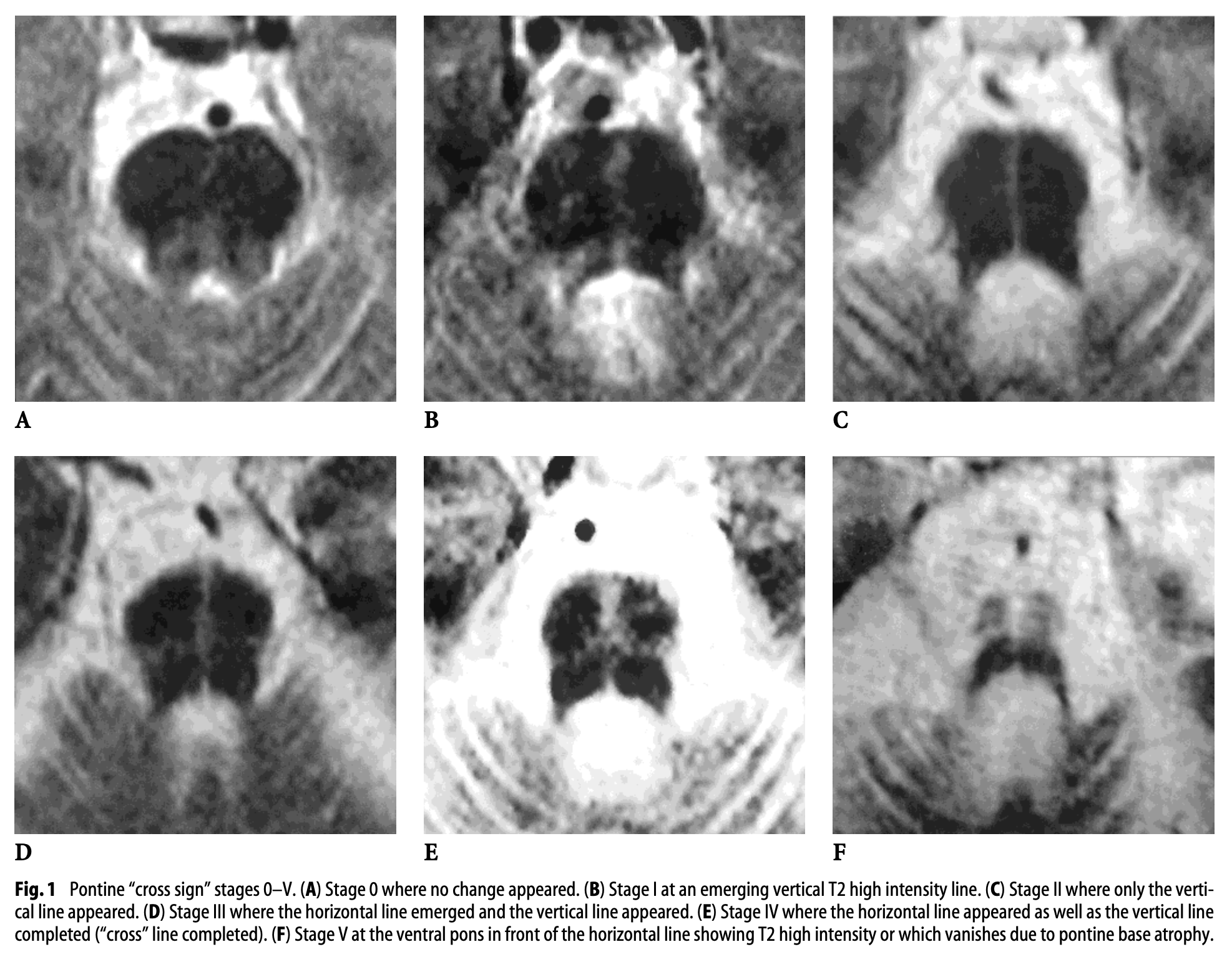
- The “hot cross buns” sign, which is a cruciform T2 signal hyperintensity in the pons.
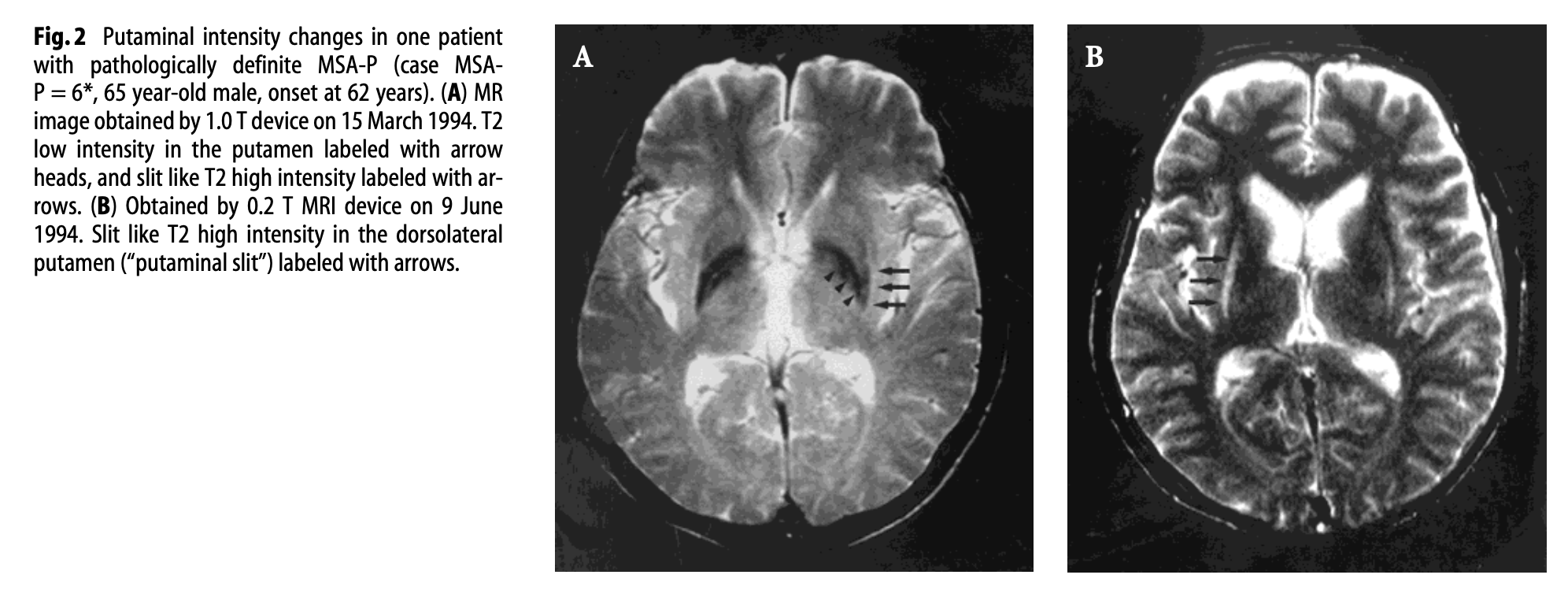
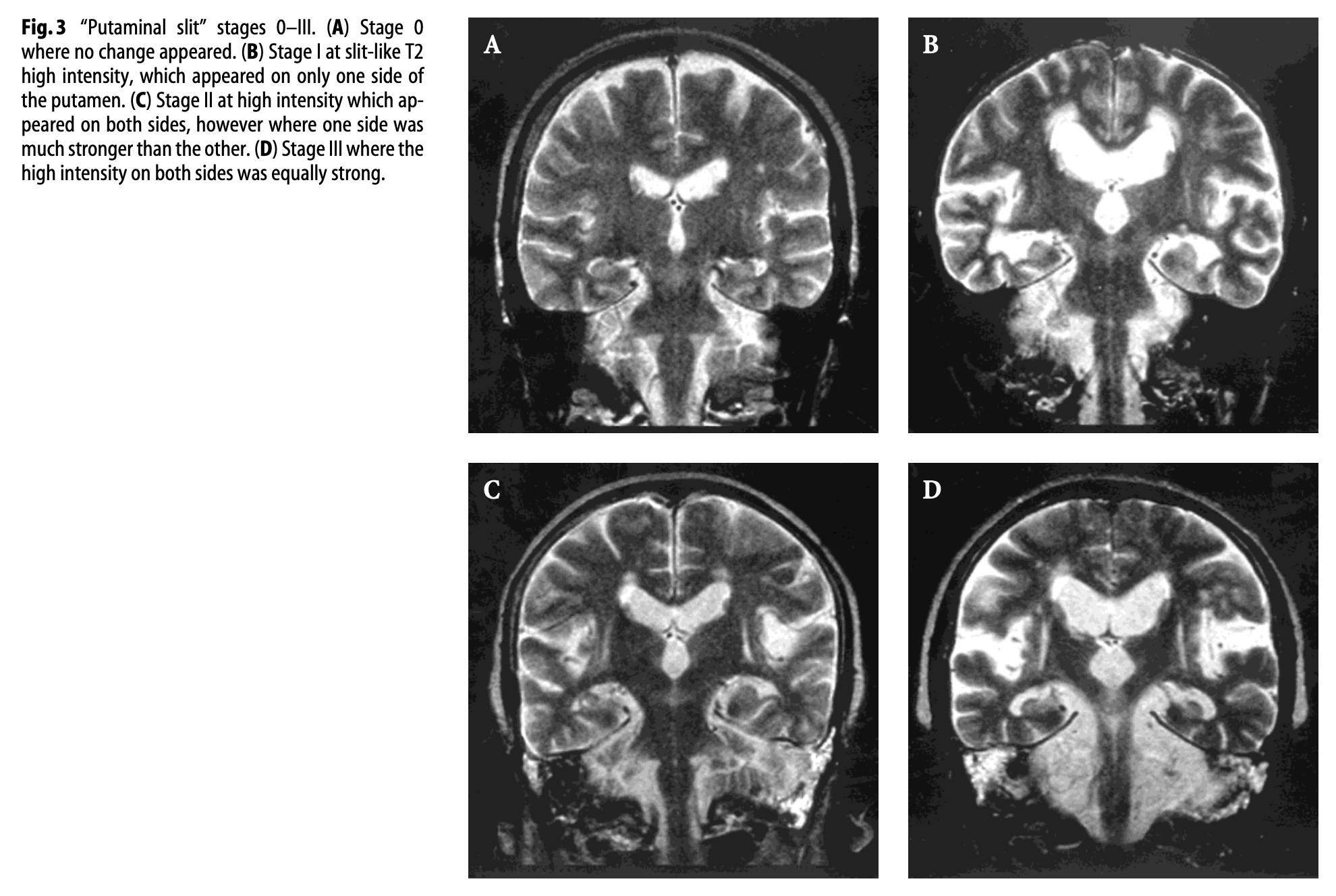
- The “putaminal slit” sign, which is a linear T2 signal hyperintensity at the dorsolateral margin of the putamen.
The Figure below, from Horimoto and colleagues (Horimoto, Aiba et al. 2002), shows evolution of the “hot cross buns” sign on serial MRIs in a patient with MSA.
The Figure below, from Horimoto and colleagues (Horimoto, Aiba et al. 2002), shows serial T2 axial MRI images demonstrating appearance of the “putaminal slit” sign in a patient with MSA.
The Figure below, from Horimoto and colleagues (Horimoto, Aiba et al. 2002), shows serial T2 coronal MRI images demonstrating appearance of the “putaminal slit” sign in a patient with MSA.
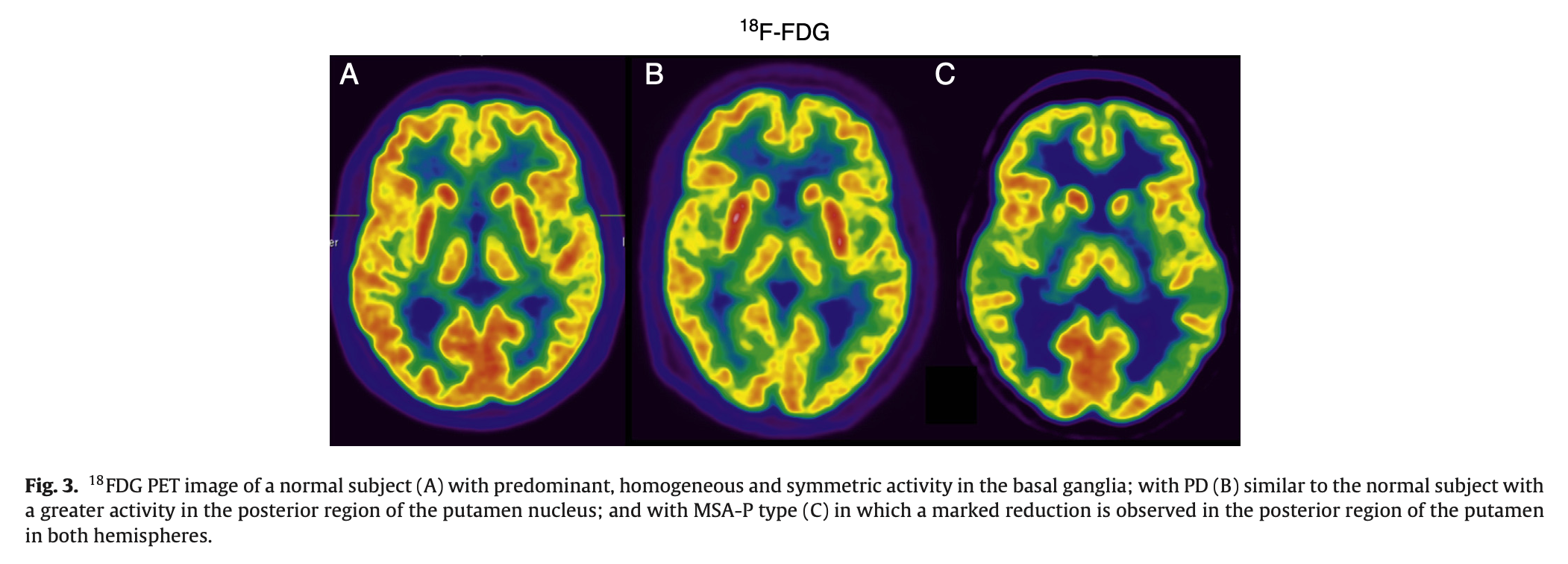
Positron emission tomography (PET) with the radionuclide tracer 18F‑fluorodeoxyglucose can reportedly differentiate MSA‑P from idiopathic Parkinson’s disease (IPD) with 95% sensitivity and 100% specificity. On this study, MSA‑P patients exhibit hypometabolism in the putamen, whereas IPD patients exhibit hypermetabolism (Arbizu, Luquin et al. 2014).
The Figure below, from Arbizu and colleagues (Arbizu, Luquin et al. 2014), compares 18F‑fluorodeoxyglucose PET scans from a healthy control (panel A), a patient with idiopathic Parkinson’s disease (panel B) and a patient with MSA‑P (panel C). The patient with MSA‑P exhibits hypometabolism in the putamina.
Histopathology
A diagnosis of MSA may be suspected antemortem, but currently a definitive diagnosis relies on post-mortem examination of the brain (Gilman, Wenning et al. 2008).
The predominant histopathological feature of MSA is the presence of glial cytoplasmic inclusions (Papp, Kahn et al. 1989), the main component of which is alpha synuclein (Spillantini, Crowther et al. 1998). Other alpha synucleinopathies include idiopathic Parkinson’s disease and Lewy body disease. MSA’s glial cytoplasmic inclusions can involve smaller amounts of other abnormal proteins as well, such as ubiquitin, tau, LRRK2, DJ‑1, p25α, GFAP and MBP (Fanciulli, Stankovic et al. 2019).
Lesions involve neuronal loss in the striatonigral and olivopontocerebellar systems, but can involve other areas in the central nervous system, peripheral nervous system and autonomic nervous system (Ubhi, Low et al. 2011). The variability in distribution and severity of this neuropathology likely accounts for the variable clinical manifestations (Fanciulli and Wenning 2015).
Differential diagnosis
The differential diagnosis includes other parkinsonian syndromes (idiopathic Parkinson’s disease, progressive supranuclear palsy, corticobasal ganglionic degeneration, Lewy body disease) and ataxias.
The Table below, from Gilman and colleagues (Gilman, Wenning et al. 2008), lists criteria for probable MSA.

The Table below, from Gilman and colleagues (Gilman, Wenning et al. 2008), lists criteria for possible MSA.

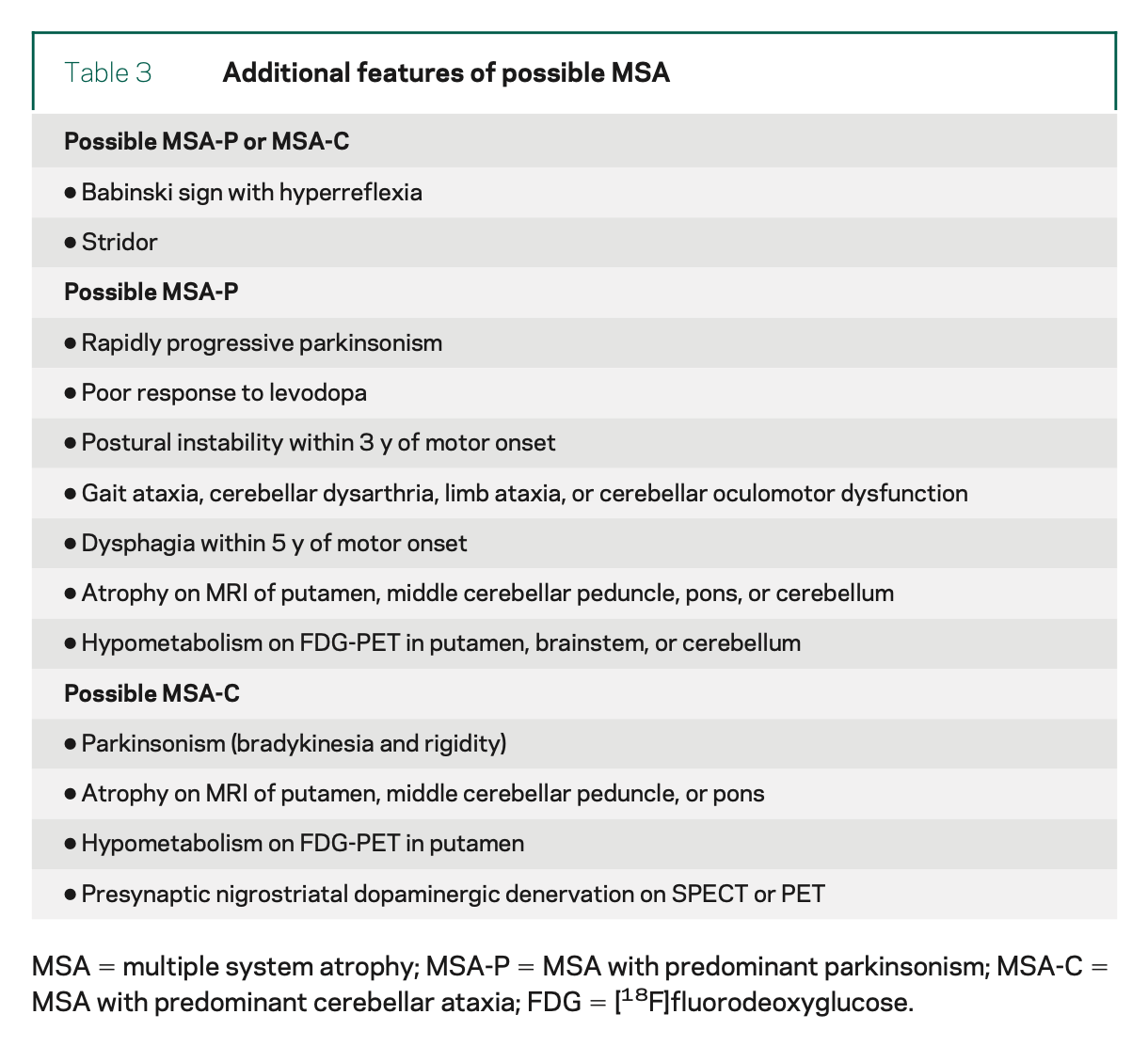
The Table below, from Gilman and colleagues (Gilman, Wenning et al. 2008), lists additional features that can support a diagnosis of MSA.
Fanciulli and colleagues (Gilman, Wenning et al. 2008) reviewed a broad range of possible biomarkers studied for MSA. The majority are of “suboptimal diagnostic accuracy” or show “insufficient evidence” of diagnostic power.
Treatment
Some MSA patient exhibit an initial, modest and transient response to levodopa treatment, which is often attempted when parkinsonian features are more prominent. This response is never sustained.
Multiple clinical trials of different interventions have been shown not to alter the disease course (Fanciulli, Stankovic et al. 2019, Meissner, Fernagut et al. 2019). These have included trials of agents with neuroprotective potential, such as human growth hormone (Holmberg, Johansson et al. 2007), minocycline (Dodel, Spottke et al. 2010), riluzole (Bensimon, Ludolph et al. 2009), rifampicin (Low, Robertson et al. 2014), rasagiline (Poewe, Seppi et al. 2015) and epigallocatechin-gallate (Levin, Maass et al. 2019).
Autologous mesenchymal stem cell administration was studied in a randomized, placebo-controlled trial (Lee, Lee et al. 2012) and an open-label trial (Singer, Berini et al. 2017), and seemed modestly to attenuate clinical progression, but remains experimental.
Since there is no curative or arrestive treatment for MSA, management strategies are symptomatic. Patients with dystonia are sometimes offered treatment with onabotulinum toxin. Physical therapy may play some role in managing parkinsonian, cerebellar and dystonic features. Autonomic features sometimes respond to pharmacotherapy (such as anticholinergics for urinary incontinence; sildenafil for erectile dysfunction; orthostatic hypotension may respond to midodrine).
Given that MSA by definition involves multiple body systems, management generally entails multiple subspecialists, which depending on the patient’s needs may be some combination of neurology, psychiatry (for mood disorders), otolaryngology (for stridor), pulmonology, sleep medicine (for REM sleep behavior disorder), cardiology (for orthostatic hypotension), gastroenterology (for constipation), urology (for neurogenic bladder), physical and occupational therapy.
Prognosis
MSA is a uniformly fatal disease (Fanciulli and Wenning 2015, Fanciulli, Stankovic et al. 2019). The mean survival from symptom onset is 9.8 years (Low, Reich et al. 2015), with a range of 6 – 10 years (Fanciulli and Wenning 2015, Meissner, Fernagut et al. 2019). Few patients survive beyond 15 years; Petrovic and colleagues (Petrovic, Ling et al. 2012) studied 135 MSA cases and found that 4 (3%) survived ≥15 years beyond symptom onset.
References
Adams RD, Vanbogaert L, Vandereecken H (1964) Striato-Nigral Degeneration. J Neuropathol Exp Neurol 23: 584-608.
Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, Holton JL (2012) The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 38: 4-24. doi: 10.1111/j.1365-2990.2011.01234.x
Anderson T, Luxon L, Quinn N, Daniel S, David Marsden C, Bronstein A (2008) Oculomotor function in multiple system atrophy: clinical and laboratory features in 30 patients. Mov Disord 23: 977-984. doi: 10.1002/mds.21999
Arbizu J, Luquin MR, Abella J, de la Fuente-Fernández R, Fernandez-Torrón R, García-Solís D, Garrastachu P, Jiménez-Hoyuela JM, Llaneza M, Lomeña F, Lorenzo-Bosquet C, Martí MJ, Martinez-Castrillo JC, Mir P, Mitjavila M, Ruiz-Martínez J, Vela L (2014) Functional neuroimaging in the diagnosis of patients with parkinsonism: Update and recommendations for clinical use. Revista Española de Medicina Nuclear e Imagen Molecular (English Edition) 33: 215-226. doi: https://doi.org/10.1016/j.remnie.2014.05.002
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN, Group NS (2009) Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 132: 156-71. doi: 10.1093/brain/awn291
Brooks SH, Klier EM, Red SD, Mehta ND, Patel SS, Chuang AZ, Suescun J, Schiess MC, Sereno AB (2017) Slowed Prosaccades and Increased Antisaccade Errors As a Potential Behavioral Biomarker of Multiple System Atrophy. Front Neurol 8: 261. doi: 10.3389/fneur.2017.00261
Coon EA, Fealey RD, Sletten DM, Mandrekar JN, Benarroch EE, Sandroni P, Low PA, Singer W (2017) Anhidrosis in multiple system atrophy involves pre- and postganglionic sudomotor dysfunction. Mov Disord 32: 397-404. doi: 10.1002/mds.26864
Dejerine JJ, Thomas A (1900) L’atrophie olivo-ponto-cérébelleuse. Nouvelle iconographie de la Salpêtriere 13: 330-370.
Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N, Trenkwalder C, Sixel-Doring F, Herting B, Kamm C, Gasser T, Sawires M, Geser F, Kollensperger M, Seppi K, Kloss M, Krause M, Daniels C, Deuschl G, Bottger S, Naumann M, Lipp A, Gruber D, Kupsch A, Du Y, Turkheimer F, Brooks DJ, Klockgether T, Poewe W, Wenning G, Schade-Brittinger C, Oertel WH, Eggert K (2010) Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord 25: 97-107. doi: 10.1002/mds.22732
Donadio V, Nolano M, Elam M, Montagna P, Provitera V, Bugiardini E, Baruzzi A, Santoro L, Liguori R (2008) Anhidrosis in multiple system atrophy: a preganglionic sudomotor dysfunction? Mov Disord 23: 885-8. doi: 10.1002/mds.21972
Doppler K, Weis J, Karl K, Ebert S, Ebentheuer J, Trenkwalder C, Klebe S, Volkmann J, Sommer C (2015) Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy. Mov Disord 30: 1688-92. doi: 10.1002/mds.26293
Fanciulli A, Stankovic I, Krismer F, Seppi K, Levin J, Wenning GK (2019) Multiple system atrophy. Int Rev Neurobiol 149: 137-192. doi: 10.1016/bs.irn.2019.10.004
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372: 249-63. doi: 10.1056/NEJMra1311488
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71: 670-6. doi: 10.1212/01.wnl.0000324625.00404.15
Graham JG, Oppenheimer DR (1969) Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry 32: 28-34. doi: 10.1136/jnnp.32.1.28
Holmberg B, Johansson JO, Poewe W, Wenning G, Quinn NP, Mathias C, Tolosa E, Cardozo A, Dizdar N, Rascol O, Slaoui T, Growth-Hormone MSASG, European MSASG (2007) Safety and tolerability of growth hormone therapy in multiple system atrophy: a double-blind, placebo-controlled study. Mov Disord 22: 1138-44. doi: 10.1002/mds.21501
Horimoto Y, Aiba I, Yasuda T, Ohkawa Y, Katayama T, Yokokawa Y, Goto A, Ito Y (2002) Longitudinal MRI study of multiple system atrophy – when do the findings appear, and what is the course? J Neurol 249: 847-54. doi: 10.1007/s00415-002-0734-0
Iodice V, Lipp A, Ahlskog JE, Sandroni P, Fealey RD, Parisi JE, Matsumoto JY, Benarroch EE, Kimpinski K, Singer W, Gehrking TL, Gehrking JA, Sletten DM, Schmeichel AM, Bower JH, Gilman S, Figueroa J, Low PA (2012) Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry 83: 453-9. doi: 10.1136/jnnp-2011-301068
Iranzo A, Santamaria J, Rye DB, Valldeoriola F, Marti MJ, Munoz E, Vilaseca I, Tolosa E (2005) Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology 65: 247-52. doi: 10.1212/01.wnl.0000168864.97813.e0
Jellinger KA (2018) Multiple System Atrophy: An Oligodendroglioneural Synucleinopathy. Journal of Alzheimer’s Disease 62: 1141-1179. doi: 10.3233/JAD-170397
Jellinger KA (2022) Heterogeneity of Multiple System Atrophy: An Update. Biomedicines 10. doi: 10.3390/biomedicines10030599
Kim HJ, Jeon BS, Lee JY, Yun JY, Kim YE, Paek SH (2012) Young-onset multiple system atrophy. J Neurol Sci 319: 168-70. doi: 10.1016/j.jns.2012.04.016
Klotz L, Klockgether T (2005) Multiple system atrophy with macrosquare-wave jerks. Mov Disord 20: 253-4. doi: 10.1002/mds.20298
Lee PH, Lee JE, Kim HS, Song SK, Lee HS, Nam HS, Cheong JW, Jeong Y, Park HJ, Kim DJ, Nam CM, Lee JD, Kim HO, Sohn YH (2012) A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol 72: 32-40. doi: 10.1002/ana.23612
Lee YH, Ando T, Lee JJ, Baek MS, Lyoo CH, Kim SJ, Kim M, Cho JW, Sohn YH, Katsuno M, Watanabe H, Yoshida M, Lee PH (2020) Later-Onset Multiple System Atrophy: A Multicenter Asian Study. Mov Disord 35: 1692-1693. doi: 10.1002/mds.28177
Levin J, Maass S, Schuberth M, Giese A, Oertel WH, Poewe W, Trenkwalder C, Wenning GK, Mansmann U, Sudmeyer M, Eggert K, Mollenhauer B, Lipp A, Lohle M, Classen J, Munchau A, Kassubek J, Gandor F, Berg D, Egert-Schwender S, Eberhardt C, Paul F, Botzel K, Ertl-Wagner B, Huppertz HJ, Ricard I, Hoglinger GU, Group PS (2019) Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 18: 724-735. doi: 10.1016/S1474-4422(19)30141-3
Low PA, Reich SG, Jankovic J, Shults CW, Stern MB, Novak P, Tanner CM, Gilman S, Marshall FJ, Wooten F, Racette B, Chelimsky T, Singer W, Sletten DM, Sandroni P, Mandrekar J (2015) Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 14: 710-9. doi: 10.1016/S1474-4422(15)00058-7
Low PA, Robertson D, Gilman S, Kaufmann H, Singer W, Biaggioni I, Freeman R, Perlman S, Hauser RA, Cheshire W, Lessig S, Vernino S, Mandrekar J, Dupont WD, Chelimsky T, Galpern WR (2014) Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 13: 268-75. doi: 10.1016/S1474-4422(13)70301-6
Meissner WG, Fernagut PO, Dehay B, Peran P, Traon AP, Foubert-Samier A, Lopez Cuina M, Bezard E, Tison F, Rascol O (2019) Multiple System Atrophy: Recent Developments and Future Perspectives. Mov Disord 34: 1629-1642. doi: 10.1002/mds.27894
Monzio Compagnoni G, Di Fonzo A (2019) Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives. Acta Neuropathol Commun 7: 113. doi: 10.1186/s40478-019-0730-6
O’Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, Revesz T, Lees AJ (2008) Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain 131: 1362-72. doi: 10.1093/brain/awn065
Palma JA, Norcliffe-Kaufmann L, Kaufmann H (2018) Diagnosis of multiple system atrophy. Auton Neurosci 211: 15-25. doi: 10.1016/j.autneu.2017.10.007
Papp MI, Kahn JE, Lantos PL (1989) Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 94: 79-100. doi: 10.1016/0022-510x(89)90219-0
Petrovic IN, Ling H, Asi Y, Ahmed Z, Kukkle PL, Hazrati LN, Lang AE, Revesz T, Holton JL, Lees AJ (2012) Multiple system atrophy-parkinsonism with slow progression and prolonged survival: a diagnostic catch. Mov Disord 27: 1186-90. doi: 10.1002/mds.25115
Pinkhardt EH, Kassubek J, Sussmuth S, Ludolph AC, Becker W, Jurgens R (2009) Comparison of smooth pursuit eye movement deficits in multiple system atrophy and Parkinson’s disease. J Neurol 256: 1438-46. doi: 10.1007/s00415-009-5131-5
Pinnock RA, McGivern RC, Forbes R, Gibson JM (2010) An exploration of ocular fixation in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. J Neurol 257: 533-9. doi: 10.1007/s00415-009-5356-3
Poewe W, Seppi K, Fitzer-Attas CJ, Wenning GK, Gilman S, Low PA, Giladi N, Barone P, Sampaio C, Eyal E, Rascol O, Rasagiline-for MSAi (2015) Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol 14: 145-52. doi: 10.1016/S1474-4422(14)70288-1
Provitera V, Nolano M, Caporaso G, Stancanelli A, Manganelli F, Iodice R, Selim MM, De Rosa A, Lanzillo B, Pellecchia MT, De Michele G, Santoro L (2014) Postganglionic sudomotor denervation in patients with multiple system atrophy. Neurology 82: 2223-9. doi: 10.1212/WNL.0000000000000518
Scarpa A, Cassandro C, Vitale C, Ralli M, Policastro A, Barone P, Cassandro E, Pellecchia MT (2020) A comparison of auditory and vestibular dysfunction in Parkinson’s disease and Multiple System Atrophy. Parkinsonism Relat Disord 71: 51-57. doi: 10.1016/j.parkreldis.2020.01.018
Schrag A, Good CD, Miszkiel K, Morris HR, Mathias CJ, Lees AJ, Quinn NP (2000) Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology 54: 697-702. doi: 10.1212/wnl.54.3.697
Shy GM, Drager GA (1960) A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol 2: 511-27. doi: 10.1001/archneur.1960.03840110025004
Singer W, Berini SE, Sandroni P, Fealey RD, Coon EA, Suarez MD, Benarroch EE, Low PA (2017) Pure autonomic failure: Predictors of conversion to clinical CNS involvement. Neurology 88: 1129-1136. doi: 10.1212/WNL.0000000000003737
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251: 205-8. doi: 10.1016/s0304-3940(98)00504-7
Ubhi K, Low P, Masliah E (2011) Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci 34: 581-90. doi: 10.1016/j.tins.2011.08.003
Venhovens J, Meulstee J, Bloem BR, Verhagen WIM (2020) Neurovestibular Dysfunction and Falls in Parkinson’s Disease and Atypical Parkinsonism: A Prospective 1 Year Follow-Up Study. Front Neurol 11: 580285. doi: 10.3389/fneur.2020.580285
Zhou H, Sun Y, Wei L, Wang X, Jiang Y, Li F, Chen J, Sun W, Zhang L, Zhao G, Wang Z (2022) Quantitative assessment of oculomotor function by videonystagmography in multiple system atrophy. Clin Neurophysiol 141: 15-23. doi: 10.1016/j.clinph.2022.05.019
Zhou H, Wang X, Ma D, Jiang Y, Li F, Sun Y, Chen J, Sun W, Pinkhardt EH, Landwehrmeyer B, Ludolph A, Zhang L, Zhao G, Wang Z (2021) The differential diagnostic value of a battery of oculomotor evaluation in Parkinson’s Disease and Multiple System Atrophy. Brain Behav 11: e02184. doi: 10.1002/brb3.2184
![]()