By Marcello Cherchi, MD PhD
For patients
Progressive supranuclear palsy (PSP) is a neurodegenerative disorder that usually begins around age 60 with symptoms such as imbalance, difficulty swallowing or pronouncing words, and urinary incontinence. Early in the disease it may resemble Parkinson’s disease. Your doctor may recognize PSP by observing specific abnormalities on physical examination. In some cases the diagnosis may be more clear from special brain scans or studies of eye movements. PSP is usually managed by a movement disorders specialist. There is no known treatment. The disease is usually fatal in 5 – 10 years from symptom onset.
For clinicians
Overview
Progressive supranuclear palsy (PSP) is a relatively rare progressive neurodegenerative tauopathy. The mean age of onset is 63 years. The disease appears to occur sporadically, though there is suggestion of genetic mutation. The only confirmed risk factor for PSP is lower educational attainment. Patients usually present with gait problems, and eventually additional symptoms such as dysarthria, dysphagia and urinary incontinence. Several sub-types of PSP have been described. On physical examination patients usually have neck hypertonia, a “frightened” appearance of facies, convergence insufficiency, and reduced amplitude and velocity of voluntary vertical (more than horizontal) saccades, with preservation of the vestibulo-ocular reflex. Instrumented ocular motor examination may reveal square wave jerks, reduced gain on smooth pursuit, and “hang up” at end trajectory of optokinetic responses. Other otovestibular tests (vestibular evoked myogenic potentials, computerized dynamic posturography) may also be abnormal, but not in a pattern that is sensitive or specific for PSP. Brain MRI often shows midbrain atrophy. FDG-SPECT scans may aid in the diagnosis. Tau-binding radioligand PET studies remain investigational. There is no proven cure or arrestive therapy, and the disease is uniformly fatal from 5.3 – 9.7 years (mean 7.4 years) after symptom onset.
The role of otoneurology is limited in PSP. If a patient originally suspected of having idiopathic Parkinson’s disease is failing to respond to therapy, then they are sometimes referred to otoneurology. Otovestibular studies (particularly videonystagmography and rotatory chair testing) may identify some of the ocular motor abnormalities that increase suspicion for PSP. This may help the patient avoid needless trials of Parkinson’s drugs, or invasive treatments.
Usually PSP patients are managed by a movement disorders neurologist.
Introduction
Progressive supranuclear palsy (PSP) is a neurodegenerative disease often classified under the broader umbrella of “Parkinson’s plus” syndromes, as it can exhibit some parkinsonian features.
At the University of Toronto, the Canadian neurologist Dr. John Clifford Richardson (1909 – 1986) observed a series of patients with the clinical findings of this disease. He worked with two colleagues at the same institution: the Polish neuropathologist Dr. Jerzy Olszewski (1913 – 1964) and another Canadian neurologist Dr. John C. Steele (1934 – 2022). In 1963 they published the first description of this clinical entity (Richardson, Steele et al. 1963, Steele, Richardson et al. 1964), which for some time was referred to by the eponymous designation, Steele-Richardson-Olszewski syndrome, but eventually came to be known as progressive supranuclear palsy (PSP).
 |
 |
 |
PSP is relatively rare, though it has attracted disproportionate attention because it has afflicted several high-profile celebrities over the years, such as Dudley Moore and Robin Williams.
Epidemiology
PSP has a prevalence of 5 – 6 cases per 100,000 people, with a mean age of onset of 63 years (Golbe 2014, Kukkle et al. 2025).
The only confirmed risk factor for PSP is lower educational attainment (Golbe, Rubin et al. 1996, Vidal, Vidailhet et al. 2009).
Genetics
PSP generally does not exhibit obvious familial patterns. However, there is evidence that certain polymorphisms of the MAPT (microtubule associated protein tau) gene on chromosome 17q21 (which encodes the tau protein) is over-represented in PSP patients (Baker, Litvan et al. 1999). The specific mutations involve inversion, deletions and partial reinversions (Golbe 2014).
The evidence for other genetic abnormalities is weaker, but includes mutations in EIF2AK3 (the gene that encodes protein kinase R-like endoplasmic reticulum kinase) which regulates protein transcription; syntaxin-6 (STX6) which is involved in vesicular exocytosis; and MOBP (myelin oligodendrocyte basic protein) which is a component of myelin (Golbe 2014).
Pathophysiological mechanism of disease
PSP is a tauopathy (Pollock, Mirra et al. 1986), and shares this molecular feature with Alzheimer disease and Pick disease. The mechanism by which accumulation of tau protein causes leads to cell dysfunction and death is unknown, though there is some suggestion that mitochondrial failure plays a role (Albers, Augood et al. 2000).
Clinical presentation
Gait difficulty is the presenting motor symptom in about 60% of cases (Golbe 2014).
Dysarthria is present in 41% of patients within 2 years of initial symptom onset (Golbe 2014).
Dysphagia is present in 18% of patients within 2 years of initial symptom onset, and in 46% of patients within 5 years of symptom onset (Golbe 2014). Dysphagia is important to recognize because it can lead to aspiration pneumonia.
Urinary incontinence is present in 42% of patients by 3.5 years from initial symptom onset (Golbe 2014).
Several sub-types of PSP have been proposed, including:
- Richardson’s syndrome
- PSP-parkinsonism
- PSP with progressive gait freezing
- PSP-corticobasal syndrome
- PSP-speech language
- PSP with frontal presentation
- PSP with predominant cerebellar ataxia
- PSP with mixed pathology
It is unclear whether these are distinct diseases, or variants of a single underlying disease. The latter possibility is suggested by what has been noted by many investigators, namely that, “The regional distribution of tau pathology and neuronal loss is a source of pathological and, consequentially, clinical heterogeneity in PSP” (Boxer, Yu et al. 2017).
Physical examination
On physical examination a PSP patient often has neck hypertonia that may be more pronounced with distraction (such as alternately opening and closing the fists). Facies are often described as having a “frightened” appearance. Face-to-face ocular motor examination may show clear convergence insufficiency; vertical (more than horizontal) saccades may be slow and of limited excursion, but reflex vestibulo-ocular reflex movements are normal.
There have been attempts at systematizing bedside ocular motor examination with the aim of distinguishing PSP from, for example, idiopathic Parkinson’s disease (Pagonabarraga, Horta-Barba et al. 2021), but none has been widely accepted or applied.
Ocular motor examination
On video Frenzel oculography one often can observe square wave jerks (SWJ). The Figure below, from Chen and colleagues (Chen, Riley et al. 2010), compares the very low amplitude refixation saccades in a healthy individual (top panel) with the square wave jerks in a PSP patient (bottom panel).
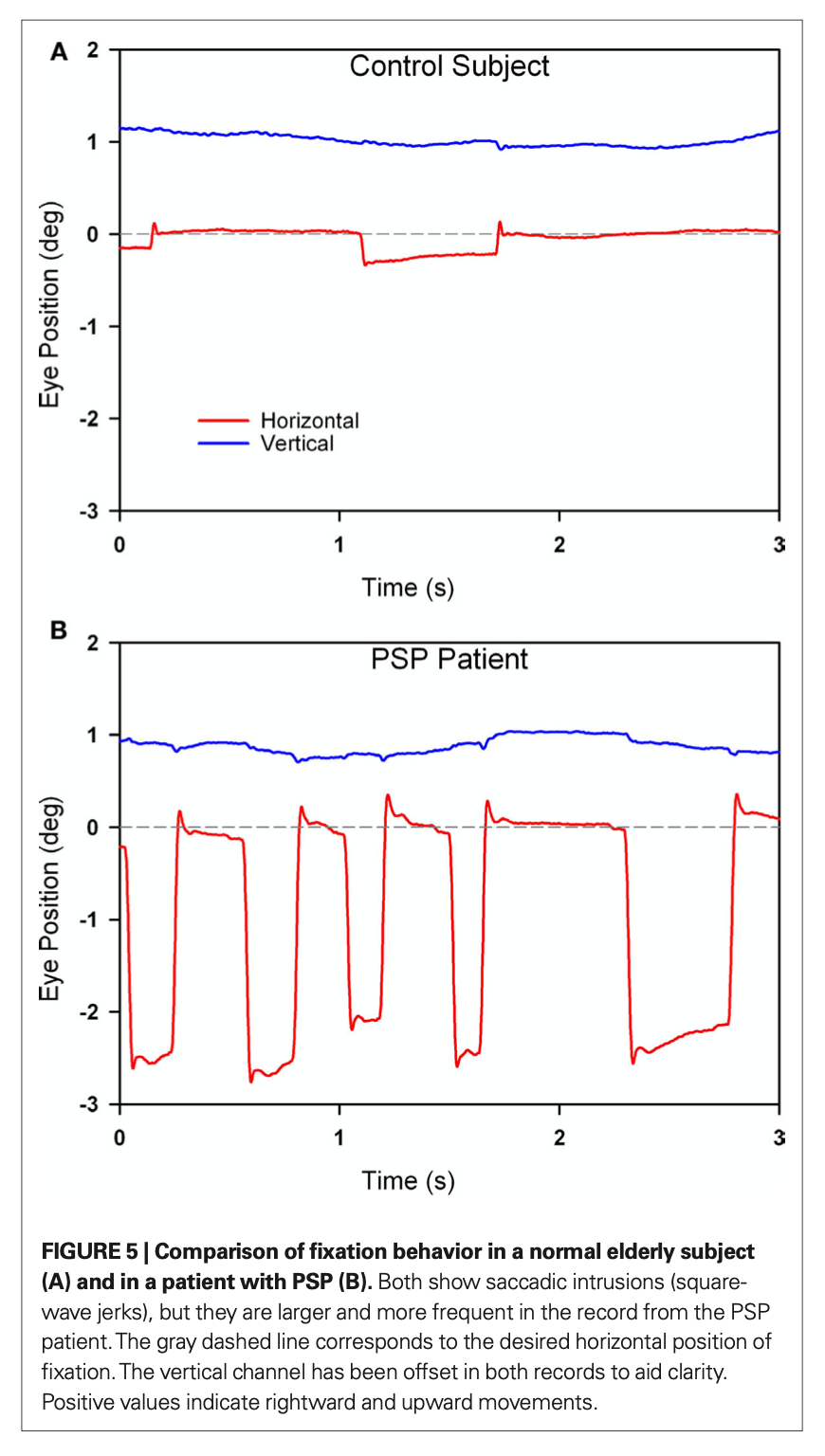
Videonystagmography (VNG) shows that vertical saccades exhibit reduced velocity and amplitude. The Figure below, from Chen and colleagues (Chen, Riley et al. 2010), compares vertical saccadic amplitude and velocity in a healthy individual (top panels) with those of a PSP patient (bottom panel).
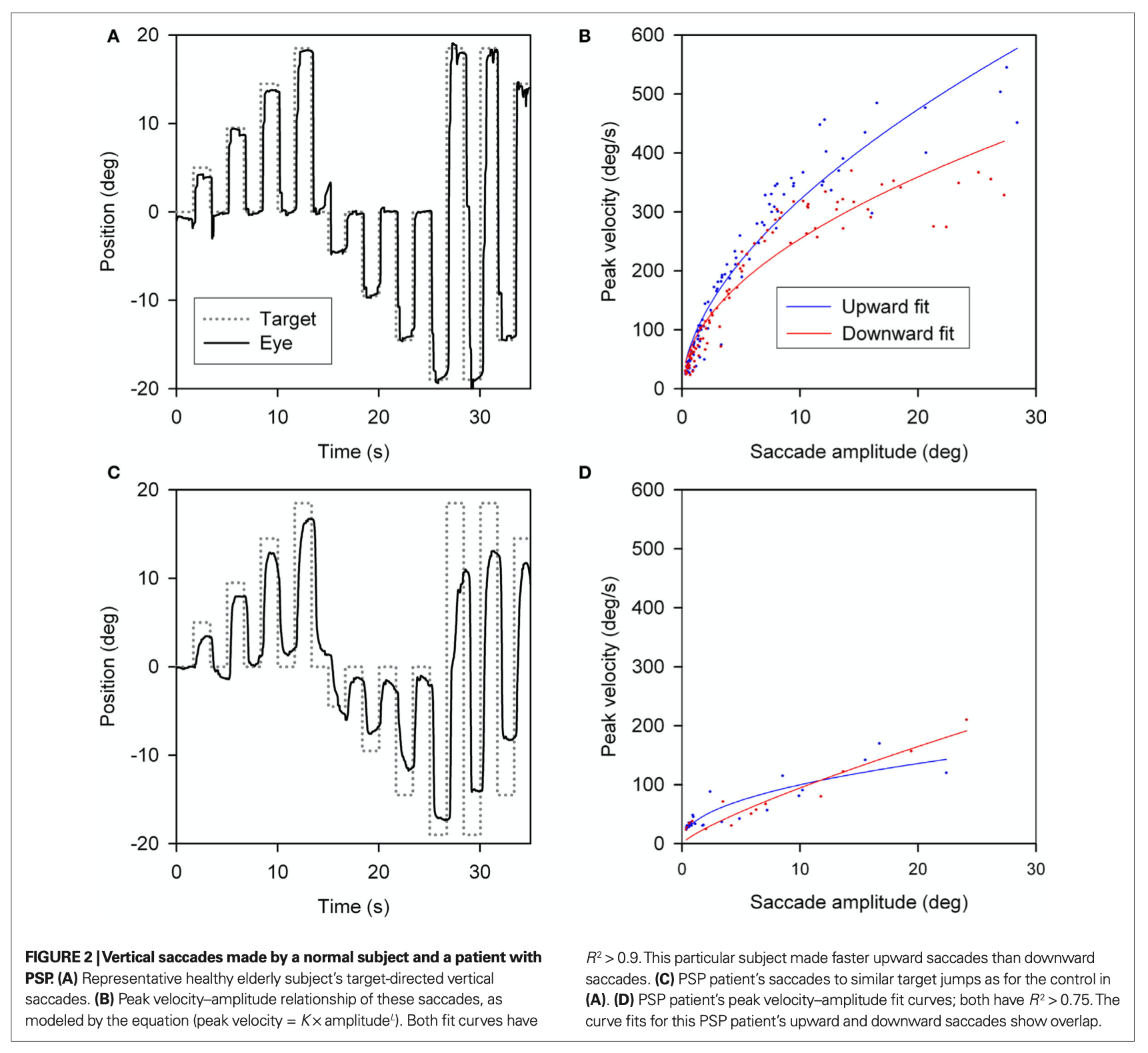
Videonystagmography (VNG) shows that vertical smooth pursuit exhibits low gain. The Figure below, from Chen and colleagues (Chen, Riley et al. 2010), compares vertical smooth pursuit in a healthy individual (top panels) with that of a PSP patient (bottom panel).
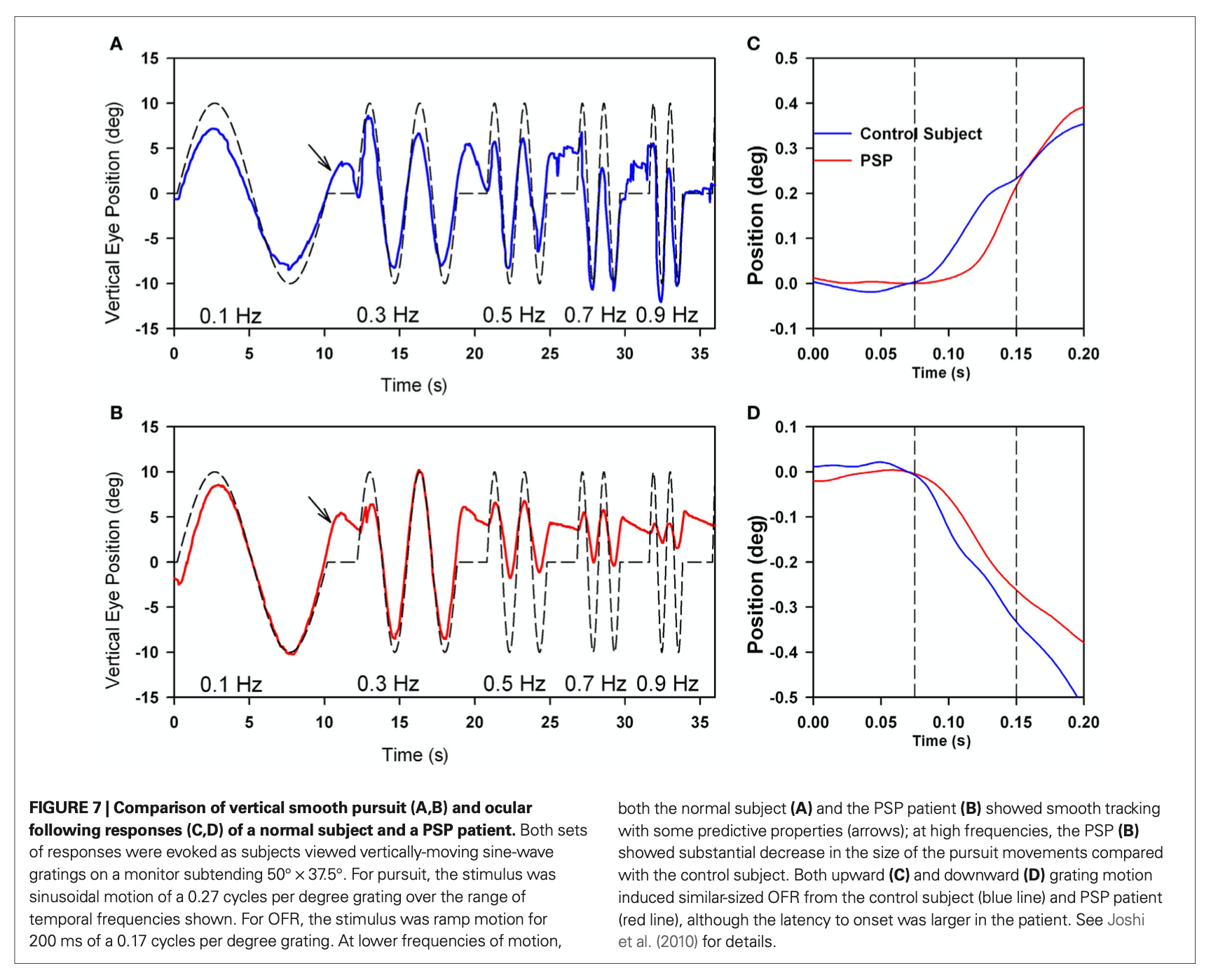
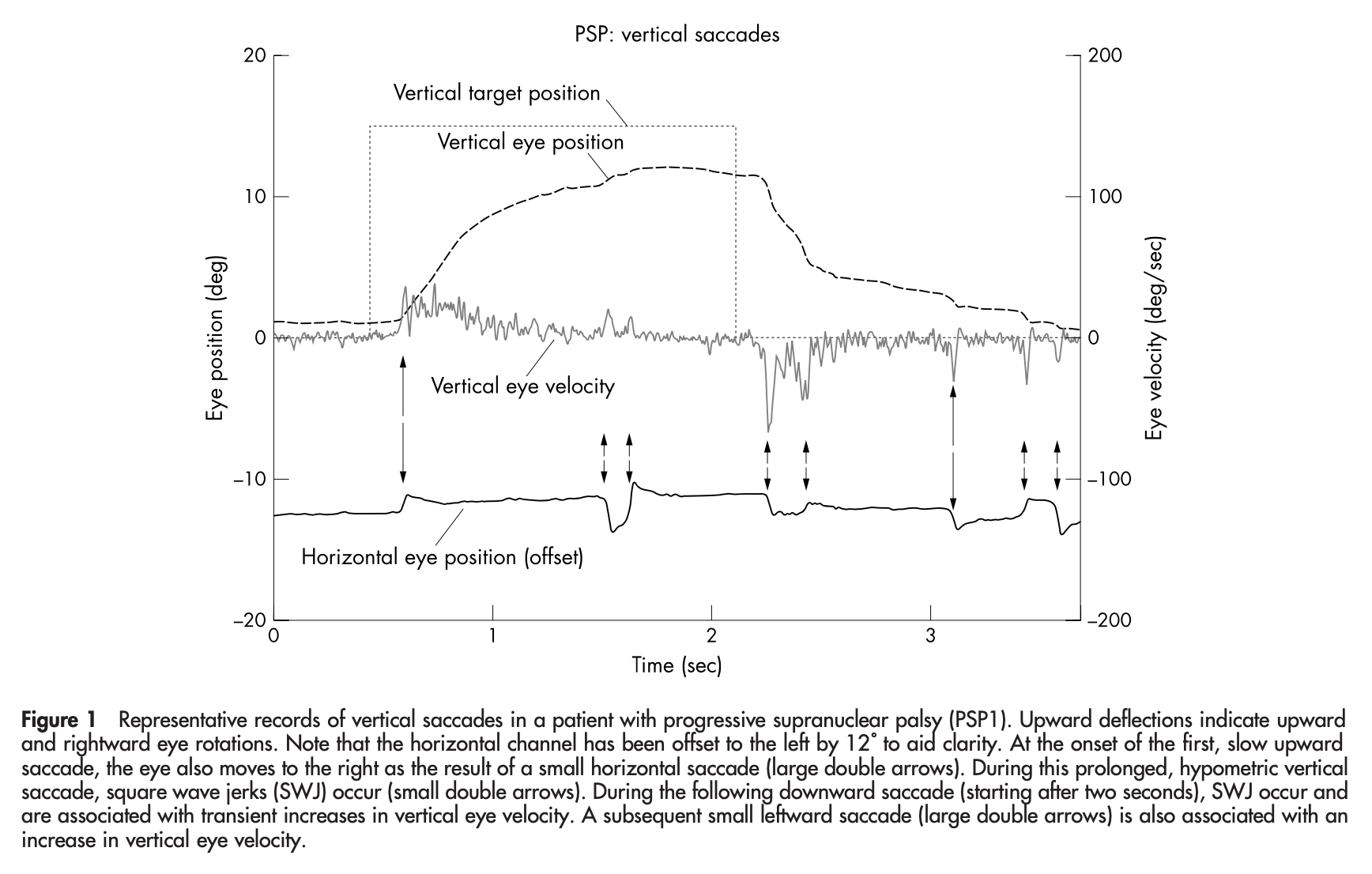
Vestibular physiologists sometimes refer to PSP as a “fast phase disorder” given the dysfunctional saccades. Since the abnormalities of saccades (reduced amplitude and velocity) are more pronounced than the gain in pursuit (reduced gain), optokinetic stimulation will tend to drag the eyes in the direction of stimulation, because the slow movement (in the direction of the optokinetic stimulus) will overwhelm the contraversive saccade (in the direction opposite that of the optokinetic stimulus), and the eyes will tend to “hang up” and the end of the trajectory. In PSP this can happen both with vertical (Garbutt, Riley et al. 2004) and horizontal optokinetic testing.
The Figure below, from Garbut and colleagues (Garbutt, Riley et al. 2004), shows “hang up” at end trajectory of vertical optokinetic responses.
Testing: vestibular evoked myogenic potentials (VEMPs)
Different studies report conflicting results on vestibular evoked myogenic potentials in PSP patients.
Carpinelli and colleagues (Carpinelli, Valko et al. 2020) reported that cervical vestibular evoked myogenic potentials (cVEMPs) and ocular vestibular evoked myogenic potentials (oVEMPs) tend to be absent more than in patients with idiopathic Parkinson’s disease, and more than in healthy controls.
In contrast, Goldschagg and colleagues (Goldschagg, Bremova-Ertl et al. 2019) report normal cVEMPs and oVEMPs in 16 PSP patients.
Testing: caloric testing
Goldschagg and colleagues (Goldschagg, Bremova-Ertl et al. 2019) studied 16 PSP patients and reported normal responses on caloric testing.
Testing: video head impulse testing (vHIT)
Goldschagg and colleagues (Goldschagg, Bremova-Ertl et al. 2019) studied 16 PSP patients and reported normal vestibulo-ocular reflex responses on video head impulse testing (vHIT).
Testing: computerized dynamic posturography (CDP)
Dale and colleagues (Dale, Horak et al. 2017) studied computerized dynamic posturography (CDP) in 12 PSP patients and reported that perception of toes up surface tilt was reduced in comparison to patients with idiopathic Parkinson’s disease and control subjects.
Goldschagg and colleagues (Goldschagg, Bremova-Ertl et al. 2019) studied CDP in 16 PSP patients and reported that the “total root-mean-square body sway was significantly increased in patients with PSP” compared to healthy control subjects.
Ondo and colleagues (Ondo, Warrior et al. 2000) studied CDP in 20 PSP patients and reported that the total limits of stability time was significantly prolonged, total path way was significantly larger, and total sensory organization testing scores were significantly worse, when compared to patients with idiopathic Parkinson’s disease.
Pasha and colleagues (Pasha, Yadav et al. 2016) studied CDP in 29 PSP patients and reported “significantly poor DP [dynamic posturography] scores” compared to healthy controls.
In summary, different CDP studies report several abnormalities in CDP, including reduced perception of toes up surface tilt, increased body sway, impaired limits of stability, and poorer sensory organization testing scores.
Imaging: MRI
It has been observed for some time that conventional MRI often shows disproportionate midbrain atrophy in PSP patients, but this finding alone is insufficiently sensitive and specific.
Josephs and colleagues (Josephs, Whitwell et al. 2008) studied 13 PSP patients; they reported brainstem atrophy and involvement of the cortex and subjacent white matter.
Massey and colleagues (Massey, Micallef et al. 2012) studied 22 PSP patients and compared them to 13 patients with multiple systems atrophy, 7 patients with idiopathic Parkinson’s disease, 6 patients with corticobasal ganglionic degeneration and 9 healthy controls. They compared conventional MRI with histopathology of each patient, and concluded that PSP patients could be distinguished from the others 73% of the time based on radiographic criteria.
Quattrone and colleagues (Quattrone, Nicoletti et al. 2008) developed what they termed the “magnetic resonance parkinsonism index (MRPI),” which consists of [pons area / midbrain area] * [middle cerebellar peduncle width / superior cerebellar peduncle width]. Their original paper attempted to distinguish PSP from idiopathic Parkinson’s disease (IPD) and from the parkinsonian variant of multiple systems atrophy (MSA‑P). The study took 33 PSP patients, 108 IPD patients, 19 MSA‑P patients and 50 healthy controls. They found that:
- In PSP patients the midbrain area and SCP width were significantly smaller than in patients with PD, MSA‑P and controls, though with some overlap.
- In PSP patients the [pons area / midbrain area] and the [middle cerebellar peduncle width / superior cerebellar peduncle width] radios were significantly larger than in patients with PD, MSA‑P and controls, though with some overlap.
- In PSP patients the MRPI (which is the [pons area / midbrain area] * [middle cerebellar peduncle width / superior cerebellar peduncle width]) was significantly larger than in patients with PD, MSA‑P and controls. They noted that the MRPI correctly identified all patients with PSP.
Hussl and colleagues (Hussl, Mahlknecht et al. 2010) also found that the MRPI was reasonably effective at distinguishing PSP from other groups (IPD, MSA‑P and healthy controls).
Morelli and colleagues (Morelli, Arabia et al. 2011) similarly found that the MRPI correctly distinguished PSP from IPD and from healthy controls. Morelli and colleagues also found the MRPI to be more accurate than clinical criteria (Morelli, Arabia et al. 2011) in predicting the development of PSP from patients with “clinically unclassifiable parkinsonism (CUP).”
Imaging: FDG-SPECT
Several studies have examined the application of fluorine-18-labelled fluorodeoxyglucose single photon emission computed tomography (FDG-SPECT) to patients with PSP.
Tang and colleagues (Tang, Poston et al. 2010) studied 167 patients diagnosed with PSP, idiopathic Parkinson’s disease and multiple systems atrophy. They found that FDG-SPECT showed 88% sensitivity, 94% specificity, 91% positive predictive value and 92% negative predictive value for PSP.
Imaging: tau-binders
Since PSP is a tauopathy, it is reasonable to consider whether tau-binding radiotracers could identify PSP patients. The ligand 18F-flortaucipir (AV‑1451) binds to paired helical filaments of tau, and several investigations have explored the utility of this radiotracer in positron emission tomography for diagnosing PSP.
A study by Whitwell and colleagues (Whitwell, Lowe et al. 2017) reported that the distribution of 18F-flortaucipir (AV‑1451) is different in PSP than in Alzheimer disease.
However, studies by Cho and colleagues (Cho, Choi et al. 2017), and by Smith and colleagues (Smith, Schain et al. 2017), did not find 18F-flortaucipir (AV‑1451) to be able to distinguish PSP from healthy controls adequately.
Currently, PET scanning remains investigational for PSP.
Histopathology
The histopathologic criteria for PSP (Litvan, Hauw et al. 1996) require findings of either neurofibrillary tangles, or tau protein neuropil threads, or both, in the basal ganglia and brainstem (Boxer, Yu et al. 2017). Other supportive features include neuronal loss, gliosis, tufted astrocytes and oligodendroglial coiled bodies (Dickson, Ahmed et al. 2010).
Differential diagnosis
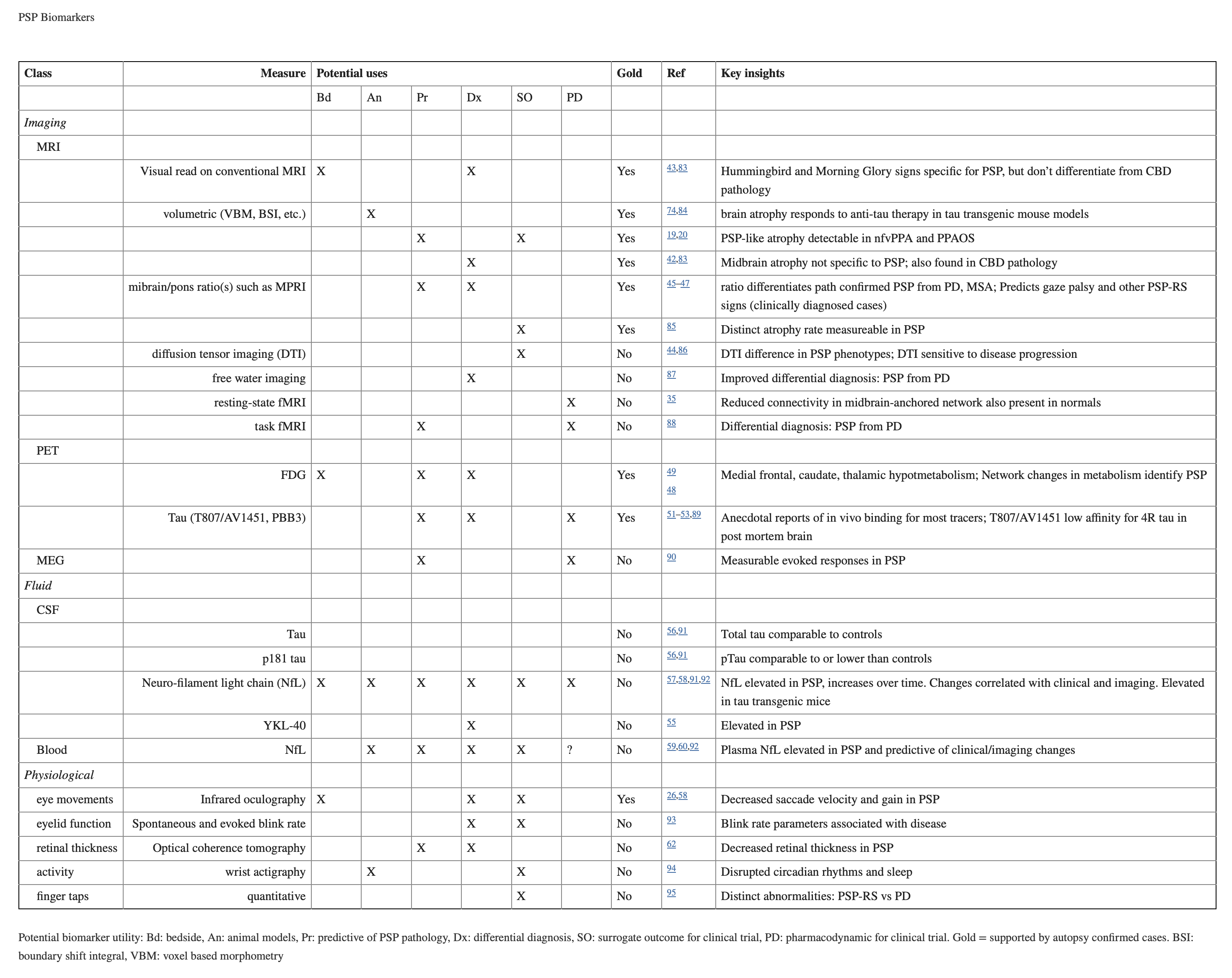
Early in the disease it can be difficult to distinguish PSP from other parkinsonian disorders. The following table, from Boxer and colleagues (Boxer, Yu et al. 2017), reviews some of the imaging, CSF, serum and physiological markers that have been explored as possible biomarkers.
Treatment
There is currently no curative or arrestive therapy for PSP. Some patients exhibit a modest and transient response to levodopa therapy. Trials of coenzyme Q10, deep brain stimulation, riluzole (Bensimon, Ludolph et al. 2009), tideglusib (Tolosa, Litvan et al. 2014) and davunetide (Boxer, Lang et al. 2014) failed to show any benefit.
Prognosis
PSP is a uniformly fatal disease. From the time of symptom onset, survival ranges from 5.3 – 9.7 years, with a mean survival of 7.4 years (Golbe 2014).
Practical summary
Progressive supranuclear palsy (PSP) is a relatively rare progressive neurodegenerative tauopathy. The mean age of onset is 63 years. The disease appears to occur sporadically, though there is suggestion of genetic mutation. The only confirmed risk factor for PSP is lower educational attainment. Patients usually present with gait problems, and eventually additional symptoms such as dysarthria, dysphagia and urinary incontinence. Several sub-types of PSP have been described. On physical examination patients usually have neck hypertonia, a “frightened” appearance of facies, convergence insufficiency, and reduced amplitude and velocity of voluntary vertical (more than horizontal) saccades, with preservation of the vestibulo-ocular reflex. Instrumented ocular motor examination may reveal square wave jerks, reduced gain on smooth pursuit, and “hang up” at end trajectory of optokinetic responses. Other otovestibular tests (vestibular evoked myogenic potentials, computerized dynamic posturography) may also be abnormal, but not in a pattern that is sensitive or specific for PSP. Brain MRI often shows midbrain atrophy. FDG-SPECT scans may aid in the diagnosis. Tau-binding radioligand PET studies remain investigational. There is no proven cure or arrestive therapy, and the disease is uniformly fatal from 5.3 – 9.7 years (mean 7.4 years) after symptom onset.
The role of otoneurology is limited in PSP. If a patient originally suspected of having idiopathic Parkinson’s disease is failing to respond to therapy, then they are sometimes referred to otoneurology. Otovestibular studies (particularly videonystagmography and rotatory chair testing) may identify some of the ocular motor abnormalities that increase suspicion for PSP. This may help the patient avoid needless trials of Parkinson’s drugs, or invasive treatments.
Usually PSP patients are managed by a movement disorders neurologist.
References
Albers DS, Augood SJ, Park LC, Browne SE, Martin DM, Adamson J, Hutton M, Standaert DG, Vonsattel JP, Gibson GE, Beal MF (2000) Frontal lobe dysfunction in progressive supranuclear palsy: evidence for oxidative stress and mitochondrial impairment. J Neurochem 74: 878-81. doi: 10.1046/j.1471-4159.2000.740878.x
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8: 711-5. doi: 10.1093/hmg/8.4.711
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN, Group NS (2009) Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 132: 156-71. doi: 10.1093/brain/awn291
Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, Doody RS, Lees A, Golbe LI, Williams DR, Corvol JC, Ludolph A, Burn D, Lorenzl S, Litvan I, Roberson ED, Hoglinger GU, Koestler M, Jack CR, Jr., Van Deerlin V, Randolph C, Lobach IV, Heuer HW, Gozes I, Parker L, Whitaker S, Hirman J, Stewart AJ, Gold M, Morimoto BH, Investigators AL (2014) Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol 13: 676-85. doi: 10.1016/S1474-4422(14)70088-2
Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Hoglinger GU (2017) Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol 16: 552-563. doi: 10.1016/S1474-4422(17)30157-6
Carpinelli S, Valko PO, Waldvogel D, Buffone E, Baumann CR, Straumann D, Werth E, Bockisch CJ, Weber KP, Valko Y (2020) Distinct Vestibular Evoked Myogenic Potentials in Patients With Parkinson Disease and Progressive Supranuclear Palsy. Front Neurol 11: 598763. doi: 10.3389/fneur.2020.598763
Chen AL, Riley DE, King SA, Joshi AC, Serra A, Liao K, Cohen ML, Otero-Millan J, Martinez-Conde S, Strupp M, Leigh RJ (2010) The disturbance of gaze in progressive supranuclear palsy: implications for pathogenesis. Front Neurol 1: 147. doi: 10.3389/fneur.2010.00147
Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS, Lyoo CH (2017) Subcortical (18) F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord 32: 134-140. doi: 10.1002/mds.26844
Dale ML, Horak FB, Wright WG, Schoneburg BM, Nutt JG, Mancini M (2017) Impaired perception of surface tilt in progressive supranuclear palsy. PLoS One 12: e0173351. doi: 10.1371/journal.pone.0173351
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23: 394-400. doi: 10.1097/WCO.0b013e32833be924
Garbutt S, Riley DE, Kumar AN, Han Y, Harwood MR, Leigh RJ (2004) Abnormalities of optokinetic nystagmus in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 75: 1386-94. doi: 10.1136/jnnp.2003.027367
Golbe LI (2014) Progressive supranuclear palsy. Semin Neurol 34: 151-9. doi: 10.1055/s-0034-1381736
Golbe LI, Rubin RS, Cody RP, Belsh JM, Duvoisin RC, Grosmann C, Lepore FE, Mark MH, Sachdeo RC, Sage JI, Zimmerman TR, Jr. (1996) Follow-up study of risk factors in progressive supranuclear palsy. Neurology 47: 148-54. doi: 10.1212/wnl.47.1.148
Goldschagg N, Bremova-Ertl T, Bardins S, Dinca N, Feil K, Krafczyk S, Lorenzl S, Strupp M (2019) No Evidence of a Contribution of the Vestibular System to Frequent Falls in Progressive Supranuclear Palsy. J Clin Neurol 15: 339-346. doi: 10.3988/jcn.2019.15.3.339
Hussl A, Mahlknecht P, Scherfler C, Esterhammer R, Schocke M, Poewe W, Seppi K (2010) Diagnostic accuracy of the magnetic resonance Parkinsonism index and the midbrain-to-pontine area ratio to differentiate progressive supranuclear palsy from Parkinson’s disease and the Parkinson variant of multiple system atrophy. Mov Disord 25: 2444-9. doi: 10.1002/mds.23351
Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Jack CR, Jr. (2008) Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 29: 280-9. doi: 10.1016/j.neurobiolaging.2006.09.019
Kukkle PL, Neupane R, Pantelyat A, Wills AM, Jabbari E, Dopper EGP, Kovacs GG, Hoglinger G, Aiba I, Litvan I, Ganguly J, Whitwell JL, Ma J, Okeng OK, Skakibara R, Forrest S, Lorenzl S, Zewde YZ, Compta Y, Morris HR, Group M-PS (2025) Progressive Supranuclear Palsy-A Global Review. Mov Disord Clin Pract. doi: 10.1002/mdc3.70338
Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55: 97-105. doi: 10.1097/00005072-199601000-00010
Massey LA, Micallef C, Paviour DC, O’Sullivan SS, Ling H, Williams DR, Kallis C, Holton JL, Revesz T, Burn DJ, Yousry T, Lees AJ, Fox NC, Jager HR (2012) Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 27: 1754-62. doi: 10.1002/mds.24968
Morelli M, Arabia G, Novellino F, Salsone M, Giofre L, Condino F, Messina D, Quattrone A (2011a) MRI measurements predict PSP in unclassifiable parkinsonisms: a cohort study. Neurology 77: 1042-7. doi: 10.1212/WNL.0b013e31822e55d0
Morelli M, Arabia G, Salsone M, Novellino F, Giofre L, Paletta R, Messina D, Nicoletti G, Condino F, Gallo O, Lanza P, Quattrone A (2011b) Accuracy of magnetic resonance parkinsonism index for differentiation of progressive supranuclear palsy from probable or possible Parkinson disease. Mov Disord 26: 527-33. doi: 10.1002/mds.23529
Ondo W, Warrior D, Overby A, Calmes J, Hendersen N, Olson S, Jankovic J (2000) Computerized posturography analysis of progressive supranuclear palsy: a case-control comparison with Parkinson’s disease and healthy controls. Arch Neurol 57: 1464-9. doi: 10.1001/archneur.57.10.1464
Pagonabarraga J, Horta-Barba A, Busteed L, Bejr-Kasem H, Illan-Gala I, Aracil-Bolanos I, Marin-Lahoz J, Pascual-Sedano B, Perez J, Campolongo A, Izquierdo C, Martinez-Horta S, Sampedro F, Kulisevsky J (2021) Quantitative evaluation of oculomotor disturbances in progressive supranuclear palsy. Parkinsonism Relat Disord 85: 63-68. doi: 10.1016/j.parkreldis.2021.03.002
Pasha SA, Yadav R, Ganeshan M, Saini J, Gupta A, Sandhya M, Pal PK (2016) Correlation between qualitative balance indices, dynamic posturography and structural brain imaging in patients with progressive supranuclear palsy and its subtypes. Neurol India 64: 633-9. doi: 10.4103/0028-3886.185417
Pollock NJ, Mirra SS, Binder LI, Hansen LA, Wood JG (1986) Filamentous aggregates in Pick’s disease, progressive supranuclear palsy, and Alzheimer’s disease share antigenic determinants with microtubule-associated protein, tau. Lancet 2: 1211. doi: 10.1016/s0140-6736(86)92212-9
Quattrone A, Nicoletti G, Messina D, Fera F, Condino F, Pugliese P, Lanza P, Barone P, Morgante L, Zappia M, Aguglia U, Gallo O (2008) MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology 246: 214-21. doi: 10.1148/radiol.2453061703
Richardson JC, Steele J, Olszewski J (1963) Supranuclear Ophthalmoplegia, Pseudobulbar Palsy, Nuchal Dystonia and Dementia. A Clinical Report on Eight Cases of “Heterogenous System Degeneration”. Trans Am Neurol Assoc 88: 25-9.
Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hagerstrom D, Jogi J, Borroni E, Scholl M, Honer M, Hansson O (2017) Increased basal ganglia binding of (18) F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord 32: 108-114. doi: 10.1002/mds.26813
Steele JC, Richardson JC, Olszewski J (1964) Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol 10: 333-59. doi: 10.1001/archneur.1964.00460160003001
Tang CC, Poston KL, Eckert T, Feigin A, Frucht S, Gudesblatt M, Dhawan V, Lesser M, Vonsattel JP, Fahn S, Eidelberg D (2010) Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol 9: 149-58. doi: 10.1016/S1474-4422(10)70002-8
Tolosa E, Litvan I, Hoglinger GU, Burn D, Lees A, Andres MV, Gomez-Carrillo B, Leon T, Del Ser T, Investigators T (2014) A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord 29: 470-8. doi: 10.1002/mds.25824
Vidal JS, Vidailhet M, Derkinderen P, de Gaillarbois TD, Tzourio C, Alperovitch A (2009) Risk factors for progressive supranuclear palsy: a case-control study in France. J Neurol Neurosurg Psychiatry 80: 1271-4. doi: 10.1136/jnnp.2008.149849
Whitwell JL, Lowe VJ, Tosakulwong N, Weigand SD, Senjem ML, Schwarz CG, Spychalla AJ, Petersen RC, Jack CR, Jr., Josephs KA (2017) [(18) F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord 32: 124-133. doi: 10.1002/mds.26834
![]()