By Marcello Cherchi, MD PhD
For patients
Usher syndrome is a hereditary cause of deafness, blindness and (sometimes) disequilibrium, with symptoms often beginning in childhood. There are several types of Usher syndrome, each with somewhat different features. The diagnosis may be suspected in someone with young and/or progressive bilateral visual impairment and bilateral hearing loss. Testing of hearing, vision and balance may further raise suspicion for the diagnosis. Confirmation of the diagnosis through genetic testing is usually offered. Depending on the type of Usher syndrome, the hearing loss may be treated with a hearing aid or cochlear implantation. Patients with disequilibrium may consider a trial of vestibular rehabilitation therapy.
For clinicians
Overview
Usher syndrome is an autosomal recessive syndrome that comprises the most common genetic cause of combined deafness and blindness, and the second most common cause of inherited hearing loss. The prevalence ranges from 1 to 10 per 30,000 individuals. There are three broad types of Usher syndrome, with multiple sub-types. Type 1 (second most common) presents with congenital, usually severe hearing loss, early visual loss, and vestibular impairment. Type 2 (most common) presents with less severe hearing loss and visual loss (though these deficits may be progressive), and normal or nearly normal vestibular function. Type 3 is variable, can be progressive, and can resemble type 1 or 2. The hearing loss is sensorineural and approximately symmetrical. Tests of vestibular function may show caloric weakness, low gain on rotatory chair testing, and impaired performance on computerized dynamic posturography. Suspected cases should be offered genetic counseling and testing. The hearing loss in Usher syndrome type 1 is often treated with cochlear implantation, while in types 2 and 3 conventional hearing aids may be attempted first. The visual loss has no known treatment, though visual accommodations can be put into place. Vestibular rehabilitation therapy can be offered to patients complaining of disequilibrium.
Introduction
Usher syndrome is the most common inherited cause of combined deafness and blindness, accounting for 50% of deaf-blind individuals younger than 65 years (Castiglione and Moller 2022). It is the second most common cause of inherited hearing loss after Pendred syndrome (Castiglione and Moller 2022).
History
German ophthalmologist Friedrich Wilhelm Ernst Albrecht von Graefe (1828 – 1870) was the first to publish a description of this disease (von Graefe 1858).
Scottish ophthalmologist, Charles Howard Usher (1865 – 1942) published a large series (69 cases) and identified the pattern of inheritance (Usher 1914), since which time the disease has borne his name.
Epidemiology
The prevalence of Usher syndrome has been difficult to determine. Conservative estimates are in the range of 1 to 10 per 30,000 individuals (Castiglione and Moller 2022), but it is the most common cause of combined hereditary deafness and blindness (Nisenbaum et al. 2022).
Among the population of deaf and hard-of-hearing individuals, Usher syndrome accounts for 3 – 6% of cases (Boughman, Vernon et al. 1983).
Usher syndrome type 1 is the second most common subtype; type 2 is the most common; type 3 is the least common (Castiglione and Moller 2022), around 1% overall, but is higher in specific populations, accounting for 40% of cases in Finland and 40% of cases among Ashkenazi Jews (Cohen, Bitner-Glindzicz et al. 2007).
Genetics
Usher syndrome is inherited in an autosomal recessive pattern.
So far three main categories of Usher syndrome have been identified, with 14 sub-types (Castiglione and Moller 2022).
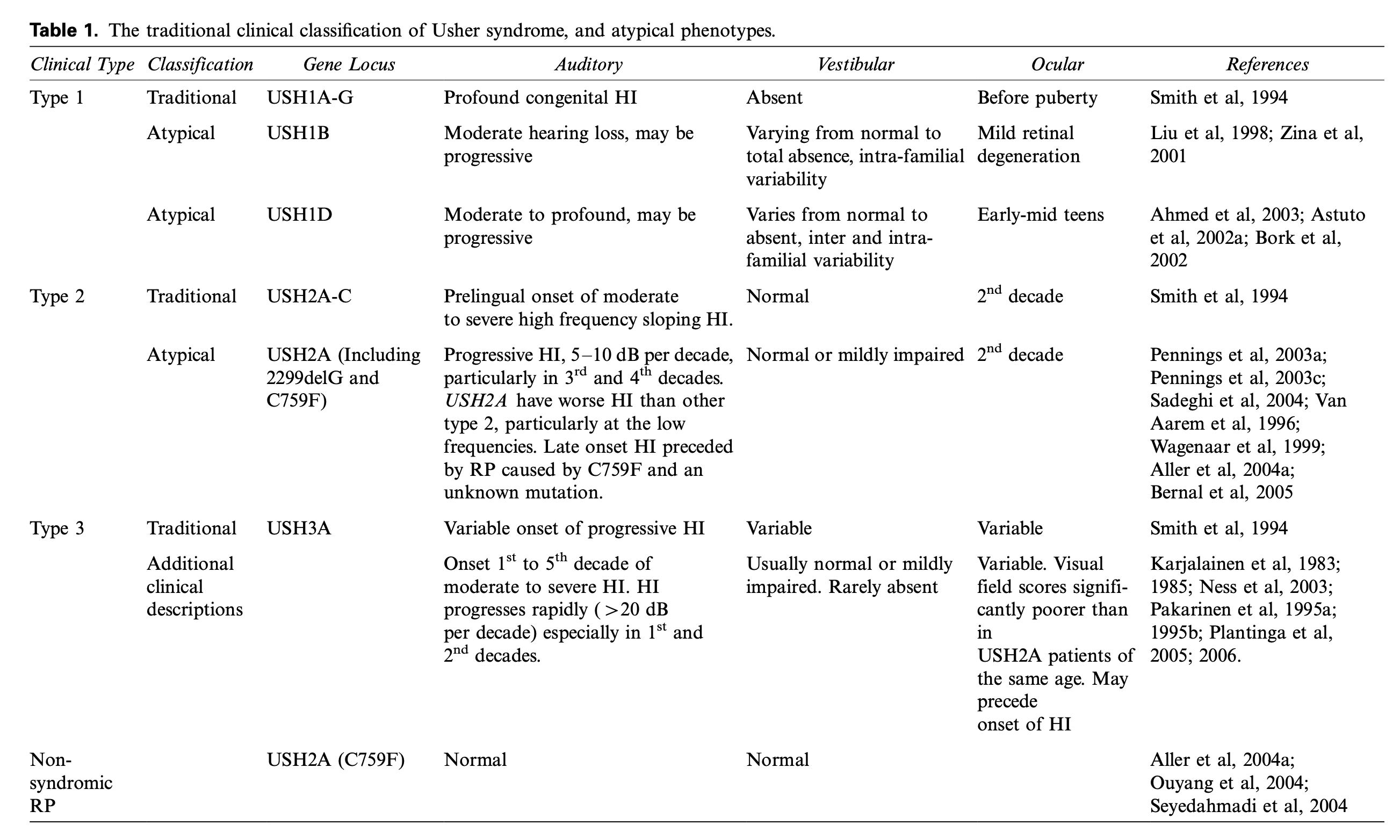
The Table below, from Cohen and colleagues (Cohen, Bitner-Glindzicz et al. 2007), is organized by type of Usher syndrome.
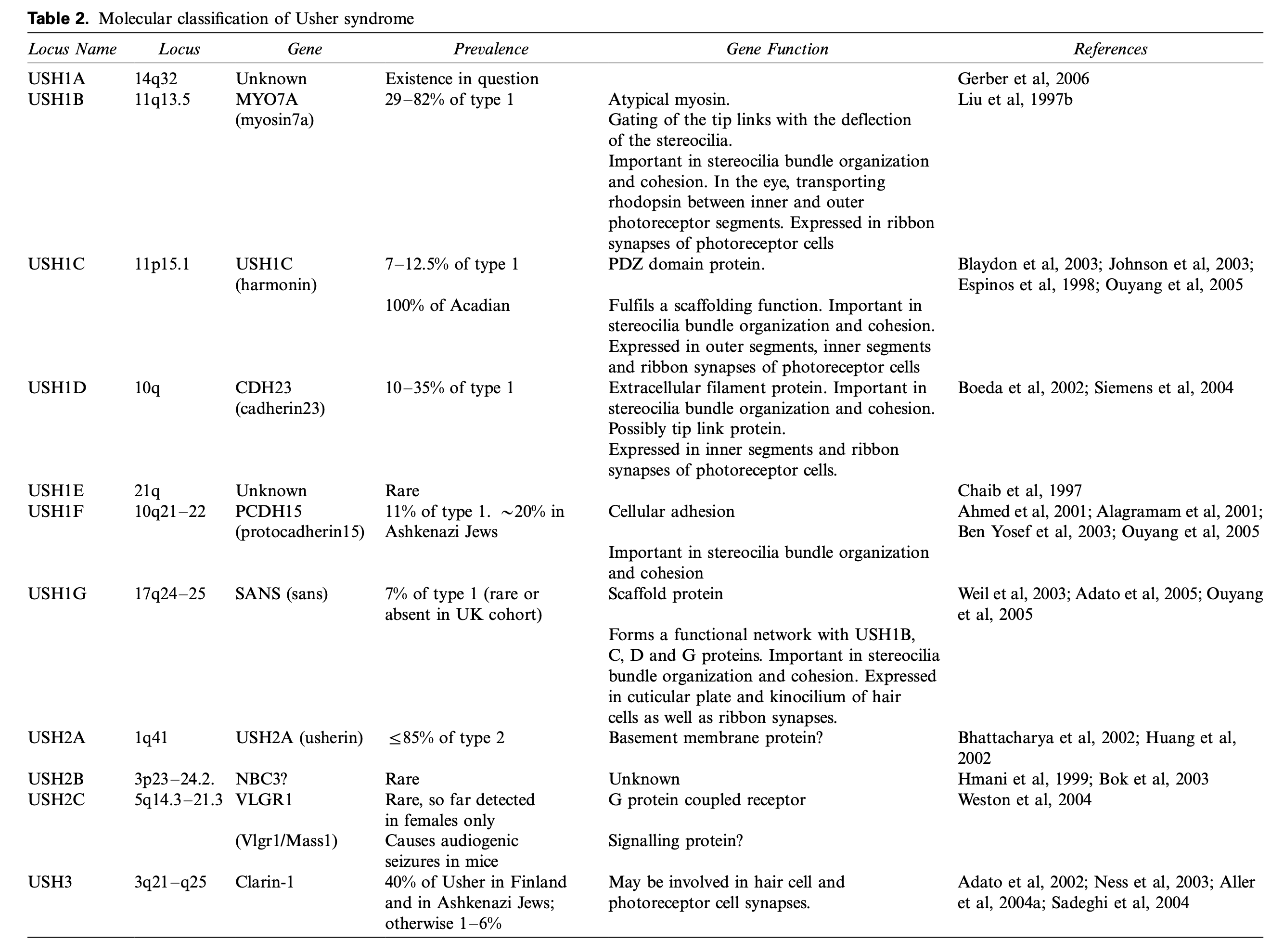
The Table below, from Cohen and colleagues (Cohen, Bitner-Glindzicz et al. 2007), is organized by molecular classification.
Pathophysiology
Usher syndrome is sometimes classified as a sensory (non-motile) ciliopathy because of its disruptive effects on genes that code for ciliary cell functions, but this is an oversimplification as it causes disruptions in other cellular functions as well (Castiglione and Moller 2022). Functionally, Usher syndrome interferes with sensory cells that express actin filaments which serve as structural proteins in hair cells and photoreceptors (Castiglione and Moller 2022). Other sensory non-motile ciliopathy syndromes include Bardet-Biedl syndrome, Alström syndrome, Meckel syndrome, Joubert syndrome and Leber congenital amaurosis (Castiglione and Moller 2022). Most investigators believe the auditory and vestibular deficits are due to end-organ (inner ear) dysfunction rather than to central (brain) dysfunction.
The Figure below, from Castiglionie and Möller (Castiglione and Moller 2022), schematically illustrates the localization in inner ear hair cells of affected proteins in the three main types of Usher syndrome.
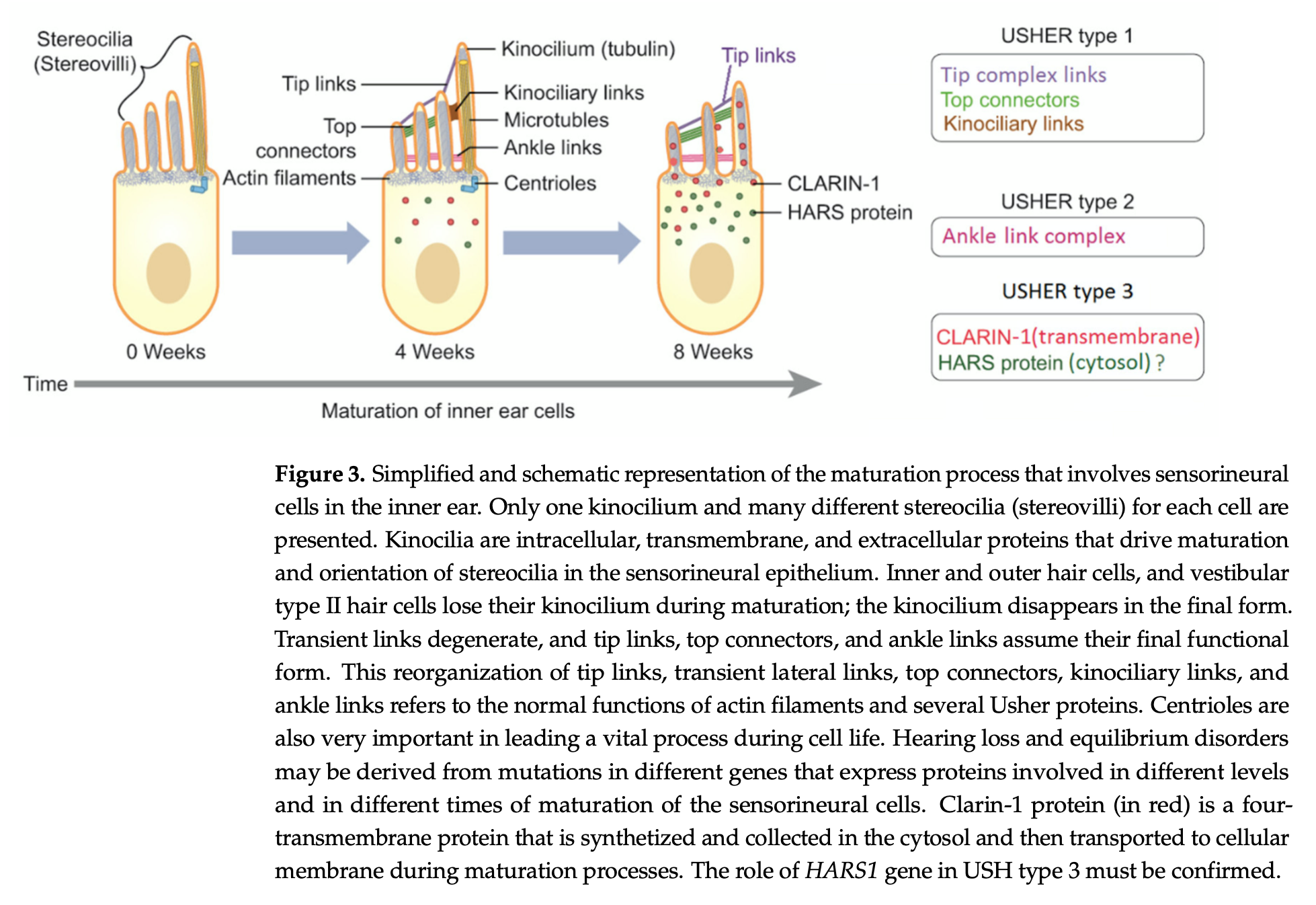
Figure schematically illustrates the localization in inner ear hair cells of affected proteins in the three main types of Usher syndrome.
Clinical presentation
The various types of Usher syndrome present with varying degrees of deficit in hearing, vision (from retinitis pigmentosa) and disequilibrium (from vestibular areflexia).
The Table below, from Castiglione and Möller (Castiglione and Moller 2022), outlines the broad patterns of hearing loss, vestibular function and visual loss, in each of the sub-types of Usher syndrome.

The Figure below, from Castiglioni and Möller (Castiglione and Moller 2022), shows general patterns of hearing loss across the three main types of Usher syndrome.
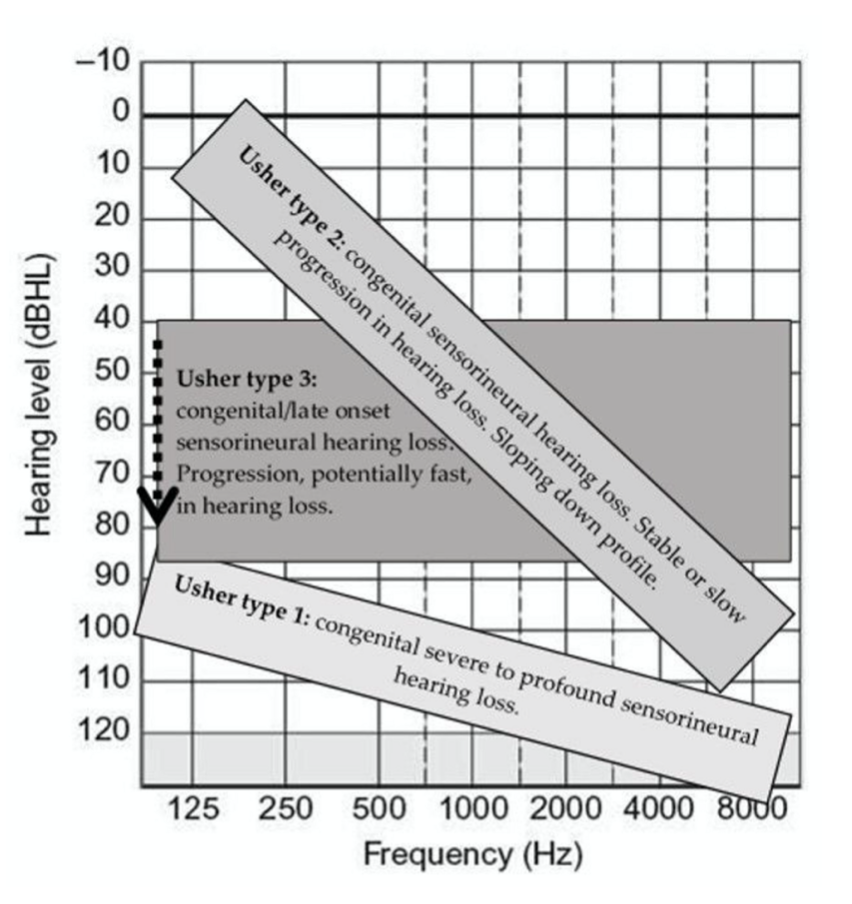
The hearing loss in all types of Usher syndrome is sensorineural and approximately symmetrical. Some researchers devote attention to statistical differences in the patterns of hearing loss, on the logic that this influences recommendations regarding amplification and cochlear implantation (Başöz Behmen et al. 2026).
The visual loss in Usher syndrome is due to retinal degeneration that begins by affecting peripheral rod cells, and gradually progresses centrally to involve cone cells (Castiglione and Moller 2022). A common initial complaint is night blindness, or generally difficulty seeing in conditions of low illumination.
The disequilibrium may manifest in early childhood as delayed motor milestones.
Workup: hearing loss
Standard audiometry shows approximately symmetrical sensorineural hearing loss that, depending on the Usher syndrome type, is variable in severity and progression.
Workup: visual loss
Retinitis pigmentosa at an early age can be confirmed by electroretinography (Castiglione and Moller 2022).
Workup: vestibular loss
Vestibular function in Usher syndrome has been studied less than hearing and vision.
Wafa and colleagues (Wafa, Faridi et al. 2021) studied 90 patients (age 10.9 – 75.5 years) from all three major types, including eight genetic subtypes, with caloric testing, rotatory chair testing, cervical vestibular evoked myogenic potentials and computerized dynamic posturography.
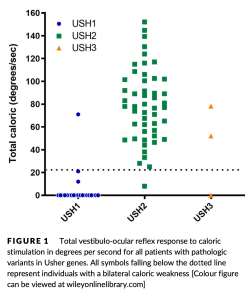
For caloric testing, Wafa and colleagues used air caloric stimulation, which seems like an odd choice since it is an even weaker stimulus than water caloric stimulation. They categorized responses as “normal, unilateral hypofunction, bilateral hypofunction, or absent.” Of 24 patients with Usher syndrome type 1, 21 (87.5%) showed no caloric response; 3 (12.5%) had some response, reflecting the overall more severe impairment in this type. Of 51 patients with Usher syndrome type 2, 45 (88%) had normal caloric responses. Of 3 patients with Usher syndrome type 3, one had bilateral caloric weakness, one had unilateral caloric weakness, and one had normal caloric responses. The overall results of caloric testing by type are displayed in the Figure below.
For rotatory chair testing, Wafa and colleagues checked slow harmonic acceleration at octave frequencies from 0.01 to 0.64 Hz and “interpreted [the responses] as absent, present with normal gain, or present with reduced gain.” Of 24 patients with Usher syndrome type 1, 14 (58.3%) had no responses. Of 49 patients with Usher syndrome type 2, 48 (96%) had normal responses. Of 6 patients with Usher syndrome type 3, responses varied from normal to absent. The overall results of slow harmonic acceleration are displayed in the Figure below.

For cervical vestibular evoked myogenic potentials, Wafa and colleagues “interpreted [the results] based on presence or absence of the P1-N1 response and interaural symmetry of the P1-N1 amplitude.” Of 20 patients with Usher syndrome type 1, 19 (95%) had bilaterally absent responses. Of 39 patients with Usher syndrome type 2, 33 (85%) had present and symmetric responses and 6 (17%) had absent responses. Of 6 patients with Usher syndrome type 3, 3 (50%) had absent responses, and 3 (50%) had present and symmetric responses.
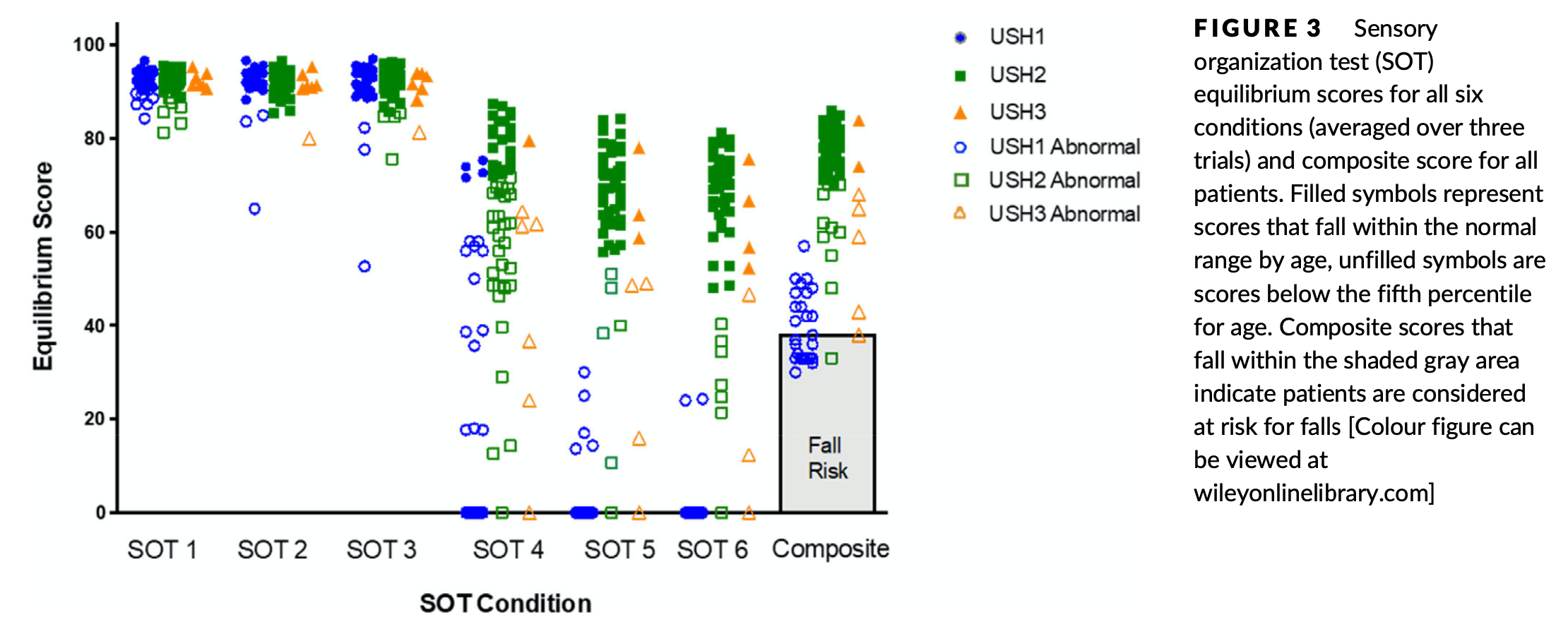
For computerized dynamic posturography, Wafa and colleagues tracked the score in each of the six conditions of sensory organization testing, as well as the composite score. Of 26 patients with Usher syndrome type 1, 21 (80%) exhibited excessive postural sway resulting in falls. Of 53 patients with Usher syndrome type 2, 48 (90%) performed normally. Of 7 patients with Usher syndrome type 3, performance was variable.
For video head impulse testing (vHIT), Amorim and colleagues (Amorim et al. 2025) note reduced rotational vestibulo-ocular reflex responses, though with differences in degree and pattern (which semicircular canals are affected) among the Usher syndrome types.
The vestibular assessments performed by Wafa and colleagues are a good starting point, but are somewhat incomplete; for example, they did not include video head impulse testing or ocular vestibular evoked myogenic potentials. Nevertheless, the overall pattern is that Usher syndrome patients of type 1 tend to have severe vestibular deficits, type 2 have modest deficits, and type 3 are variable.
As far as rotatory chair testing is concerned, other studies have noted that Usher syndrome type 1 patients have significant deficits, while type 2 patients are normal (Moller, Kimberling et al. 1989).
As far as computerized dynamic posturography is concerned, other studies have noted that Usher syndrome type 1 patients on average perform more poorly than type 2 (Moller, Kimberling et al. 1989, Caldani, Bucci et al. 2019).
The variability of vestibular impairment in Usher syndrome type 3 has been noted by other investigators (Sadeghi, Cohn et al. 2005, Marouf, Johnson et al. 2022), and in this respect type 3 can mimic types 1 and 2 (Pennings, Fields et al. 2003).
Workup: genetic testing
Patients with clinical features of hearing loss and visual loss from a young age should consider consultation with a medical geneticist regarding genetic testing for Usher (and other) syndromes.
Treatment
Usher syndrome type 1 usually presents with profound hearing loss. Since there is no serviceable hearing, cochlear implantation is often suggested. Usher syndrome types 2 and 3 may have less severe hearing loss, and these patients may benefit from a conventional hearing aid.
When the vision loss is recognized, accommodations can be made in terms of lighting and contrast conditions.
Rehabilitation for disequilibrium may attempt sensory substitution with vision (if there remains serviceable vision) and somatosensory input.
References
Amorim AM, Ramada AB, Lopes AC, Figueiredo HB, Lemos J, Ribeiro JC (2025) Vestibular Phenotype-Genotype Correlation in a Cohort of 35 European Usher Syndrome Patients. Am J Audiol 34: 502-515. doi: 10.1044/2025_AJA-24-00194
Başöz Behmen M, Yılmaz R, Tas Elibol N (2026) Audiological findings in usher syndrome according to types. The Egyptian Journal of Otolaryngology 42: 2. doi: 10.1186/s43163-025-00980-7
Boughman JA, Vernon M, Shaver KA (1983) Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis 36: 595-603. doi: 10.1016/0021-9681(83)90147-9
Caldani S, Bucci MP, Tisne M, Audo I, Van Den Abbeele T, Wiener-Vacher S (2019) Postural Instability in Subjects With Usher Syndrome. Front Neurol 10: 830. doi: 10.3389/fneur.2019.00830
Castiglione A, Moller C (2022) Usher Syndrome. Audiol Res 12: 42-65. doi: 10.3390/audiolres12010005
Cohen M, Bitner-Glindzicz M, Luxon L (2007) The changing face of Usher syndrome: clinical implications. Int J Audiol 46: 82-93. doi: 10.1080/14992020600975279
Marouf A, Johnson B, Alagramam KN (2022) Usher syndrome IIIA: a review of the disorder and preclinical research advances in therapeutic approaches. Hum Genet 141: 759-783. doi: 10.1007/s00439-022-02446-9
Moller CG, Kimberling WJ, Davenport SL, Priluck I, White V, Biscone-Halterman K, Odkvist LM, Brookhouser PE, Lund G, Grissom TJ (1989) Usher syndrome: an otoneurologic study. Laryngoscope 99: 73-9. doi: 10.1288/00005537-198901000-00014
Nisenbaum E, Thielhelm TP, Nourbakhsh A, Yan D, Blanton SH, Shu Y, Koehler KR, El-Amraoui A, Chen Z, Lam BL, Liu X (2022) Review of Genotype-Phenotype Correlations in Usher Syndrome. Ear Hear 43: 1-8. doi: 10.1097/AUD.0000000000001066
Pennings RJ, Fields RR, Huygen PL, Deutman AF, Kimberling WJ, Cremers CW (2003) Usher syndrome type III can mimic other types of Usher syndrome. Ann Otol Rhinol Laryngol 112: 525-30. doi: 10.1177/000348940311200608
Sadeghi M, Cohn ES, Kimberling WJ, Tranebjaerg L, Moller C (2005) Audiological and vestibular features in affected subjects with USH3: a genotype/phenotype correlation. Int J Audiol 44: 307-16. doi: 10.1080/14992020500060610
Usher CH (1914) On the inheritance of retinitis pigmentosa, with notes of cases. R Lond Ophthal Hosp Rep 19: 130-256.
von Graefe A (1858) Vereinzelte Beobachtungen und Bemerkungen: Exeptionelles Verhalten des Gesichtfeldes bei Pigmentartung der Netzhaut [Unique observations and remarks: Exceptional behavior of the field of vision in retinal pigmentation]. Archiv für klinische und experimentelle Ophthalmologie [Archive for Clinical and Experimental Ophthalmology] 4: 250-253.
Wafa TT, Faridi R, King KA, Zalewski C, Yousaf R, Schultz JM, Morell RJ, Muskett J, Turriff A, Tsilou E, Griffith AJ, Friedman TB, Zein WM, Brewer CC (2021) Vestibular phenotype-genotype correlation in a cohort of 90 patients with Usher syndrome. Clin Genet 99: 226-235. doi: 10.1111/cge.13868
![]()