By Marcello Cherchi, MD PhD
For patients
Refsum disease (RD) is a genetic disorder that causes an abnormal build-up of phytanic acid in the bloodstream and various organs. In late childhood or in adulthood, patients may develop symptoms such as poor vision, poor hearing, poor sense of smell, scaly skin, and others. If your doctor suspects RD, then they may check certain tests of vision, hearing, balance, and blood tests. Depending on what they discover, they may recommend that you consult with a dietician, ophthalmologist, audiologist, otolaryngologist, cardiologist or physical therapist.
For clinicians
Overview
Refsum disease (RD) is an autosomal recessive metabolic disorder of lipid metabolism that causes abnormal accumulation of phytanic acid. Depending on where the phytanic acid accumulates, patients may suffer from anosmia, vision loss (from retinitis pigmentosa), sensorineural hearing loss, imbalance (from vestibular weakness and cerebellar dysfunction), somatosensory deficits and weakness (from sensorimotor neuropathy), scaly skin (from ichthyosis) and cardiac problems (arrhythmia, cardiac failure). Workup reveals elevated serum levels of phytanic acid; genetic testing should confirm one of several possible mutations. Additional findings on a patient’s workup will depend on the specific deficits (ophthalmologic, audio-vestibular, neuromuscular, etc.). Brain MRI findings are non-specific. Treatment involves restriction of dietary phytanic acid intake; this can retard progression, and sometimes even reverse some laboratory findings.
Introduction
Refsum disease (RD) is an autosomal recessive metabolic disorder of peroxisomal lipid metabolism whose adult form results from mutations in PHYX/PAHX or PEX7 genes, and whose infantile form results from a mutations in PEX1 or PEX6 genes. These mutations impair the catabolism phytanic acid, whose dietary sources include vegetables, meat of ruminant animals (beef, lamb, veal) and some predatory fish (cod, tuna). As a result, phytanic acid gradually accumulates in various tissues, and can cause anosmia, vision loss (from retinitis pigmentosa), sensorineural hearing loss, imbalance (from vestibular weakness and cerebellar dysfunction), somatosensory deficits and weakness (from sensorimotor neuropathy), scaly skin (from ichthyosis) and cardiac problems (arrhythmia, cardiac failure).
Historical background
Sigvald Bernhard Refsum (1907 – 1991) (Nyberg-Hansen 1992) was a Norwegian neurologist. In 1946 he published the first description of a syndrome (Refsum 1946) that he described as “heredopathia atactica polyneuritiformis” (hereditary ataxia with polyneuritis). He characterized the syndrome further in several subsequent papers (Refsum 1952, 1981; Refsum et al. 1949), and it eventually came to bear his name. Hereditary motor and sensory neuropathy (HMSN) type 4 is another designation for the neuropathy that occurs in Refsum disease.
Epidemiology
Refsum disease is rare. Its precise prevalence is unknown, but estimated at less than 1 per million.
Genetics
Refsum disease (OMIM 26500) is an autosomal recessive neurocutaneous disorder of lipid metabolism that can arise from several mutations, each of which causes abnormal accumulation in plasma and various tissues of phytanic acid, a branched-chain fatty acid.
Most cases of non-infantile Refsum disease result from a mutation in a gene that encodes for the peroxisomal enzyme phytanoyl-CoA hydroxylase, which converts phytanic acid to phytanoyl-CoA by alpha oxidation. This gene’s locus is on chromosome 10p13 (OMIM 602026), and is usually referred to as PHYX (Jansen et al. 2000; Jansen et al. 1997), less commonly as PAHX (Mihalik et al. 1997). PAHX is expressed in cells of the “supragranular layer in the cerebral cortex, dentate gyrus, hippocampus, Purkinje cell layer, deep cerebellar nucleus, trigeminal nucleus, abducent nucleus, facial nucleus, cochlear and vestibular nucleus, ganglion cell and nuclear layer of the retina” (Lee et al. 2000).
A minority of cases of non-infantile Refsum disease result from a mutation in the PEX7 (peroxisome biogenesis factor 7) gene (OMIM 601757) at a locus on chromosome 6q22-24 (van den Brink et al. 2003). Peroxisome biogenesis factor 7 is required for peroxisomal import of proteins.
Infantile Refsum disease results from a mutation in PEX1 (OMIM 602136) (Portsteffen et al. 1997; Reuber et al. 1997) or PEX6 (Geisbrecht et al. 1998)
Pathophysiological mechanism of disease
Phytanic acid is an exogenous compound present in most people’s diet. The main dietary sources of phytanic acid are vegetables, dairy products, meat of ruminant animals (beef, lamb, veal) and some predatory fish (cod, tuna) (Ruether et al. 2010). The biochemical abnormalities in Refsum disease impair catabolism of phytanic acid, so phytanic acid circulates in serum and damages tissues in which it accumulates.
The mechanism of sensorineural hearing loss in Refsum disease is not clear. Taylor and colleagues report the case of a 42-year-old woman with longstanding hearing loss who had abnormal otoacoustic emissions (OAE) and nearly normal auditory brainstem evoked responses (ABR), compatible with a cochlear deficit. In contrast, Oysu and colleagues (Oysu et al. 2001) report the case of a 6-year-old boy with hearing loss in whom otoacoustic emissions (OAE) were present and auditory brainstem evoked responses (ABR) were absent, compatible with an auditory neuropathy. Similarly, Vandana and colleagues (Vandana et al. 2015) describe the case of a 4-year-old boy with hearing loss in whom otoacoustic emissions (OAE) were preserved and auditory brainstem evoked responses (ABR) were absent.
The mechanism of cerebellar dysfunction in Refsum disease is unclear (Salisachs 1982), since the few available autopsy studies report limited pathology in the cerebellum itself, though the existing studies do note “atrophy of the cerebellar vermis and patchy cell degeneration in the dentate and inferior olivary nuclei” (De Munter et al. 2015). One possibility is that the apparent cerebellar ataxia is due to neuropathically deficient afferents to the cerebellum (similar to Friedreich ataxia). In contrast, a mouse model of Refsum disease suggests that the deficit is in the cerebellum (Ferdinandusse et al. 2008).
Clinical presentation
Refsum disease can present at any point from infancy to adulthood. It usually manifests before the age of 20 years, but has been reported as late at 50 years (Ruether et al. 2010).
Accumulation of phytanic acid in various tissues results in some combination of the following features:
- Cranial and peripheral neuropathies:
- Vision loss from retinitis pigmentosa (Ruether et al. 2010), often presenting as impaired night vision.
- Anosmia (Gibberd et al. 2004).
- Sensorineural hearing loss (Bamiou et al. 2003).
- Vestibular weakness (Taylor et al. 2018).
- Sensorimotor polyneuropathy (Gelot et al. 1995).
- Ataxia from cerebellar dysfunction.
- Ophthalmologic manifestations (Claridge et al. 1992; Dick et al. 1990; Ruether et al. 2010):
- Miosis
- Iris atrophy
- Cataract
- Ichthyosis (Davies et al. 1977).
- Cardiac arrhythmias, or sometimes dilated cardiomyopathy (Arous et al. 2024), which can be a fatal complication (Allen et al. 1978).
Sensorineural hearing loss is reported to occur in up to 80% of patients (Bergsmark and Djupesland 1968; Feldmann 1981; Oysu et al. 2001).
Symptomatic exacerbations may occur under physiologically demanding circumstances (such as a concomitant illness, surgery, pregnancy, fasting, rapid weight loss, etc.).
Physical examination
Not all patients exhibit all deficits, consequently physical examination findings are variable (Dotti et al. 1985), but can include:
- Anosmia.
- Sensorineural hearing loss.
- Visual loss.
- Imbalance and ataxia.
- Evidence of sensorineural peripheral neuropathy, such as weakness and diminished myotatic reflexes.
- Ichthyosis.
Ocular motor examination
Weleber and colleagues (Weleber et al. 1984) describe a case of a patient with Refsum disease who began to exhibit “nystagmus” in “early infancy,” but they did not provide any details of the nystagmus. Tran and colleagues (Tran et al. 2011) similarly mention “nystagmus” in a 15-year-old, without providing further description.
Testing: auditory
Taylor and colleagues (Taylor et al. 2018) described the case of a 42-year-old woman with Refsum disease who reported “longstanding bilateral hearing loss.” They reported that audiometry showed “mild to moderate sensorineural hearing loss bilaterally” and “Otoacoustic emissions were mostly absent,” whereas “Auditory brainstem responses indicated preservation of all waves with borderline abnormal interpeak latencies I-III and I-V.”
Testing: vestibular
Harris and colleagues (Harris et al. 1996) report intermittent failure of horizontal saccades in a group of children (age 17 days to 14 years).
Taylor and colleagues (Taylor et al. 2018) described the case of a 42-year-old woman with Refsum disease who reported a 2-month history of disequilibrium. They reported delayed prolonged latencies of cervical vestibular evoked myogenic potentials (cVEMP) and ocular vestibular evoked myogenic potentials (oVEMP). In contrast, results of video head impulse testing (vHIT) were normal. They further noted that the vestibular evoked myogenic potentials (VEMP) responses improved as treatment reduced the serum phytanic acid levels. This improvement is shown in the Figure below.
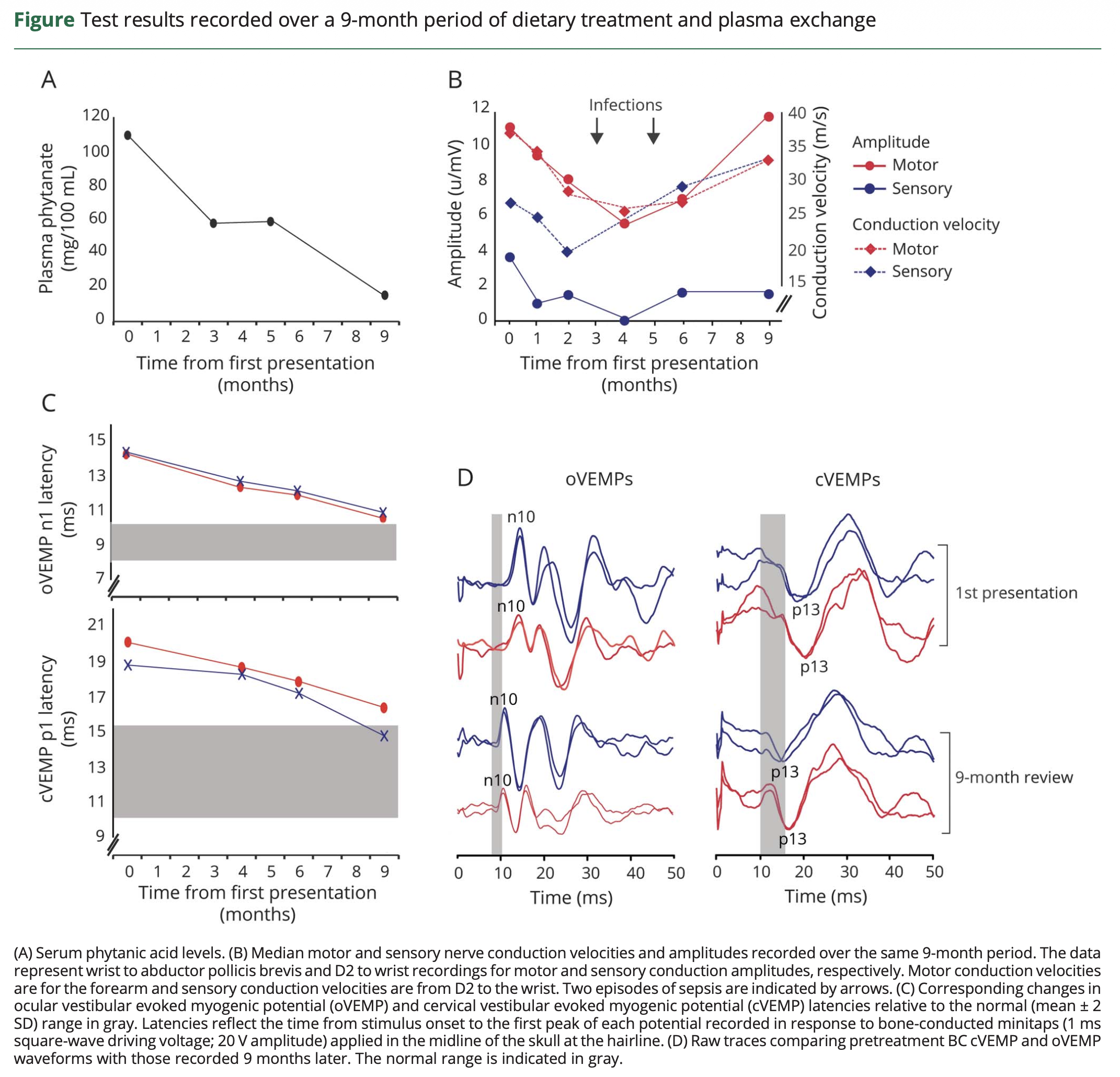
Figure : Cervical vestibular evoked myogenic potentials (cVEMP) and ocular vestibular evoked myogenic potentials (oVEMP) in a 42-year-old patient with Refsum disease. Initially the VEMP latencies were prolonged. The VEMP responses improved as treatment gradually improved the serum level of phytanic acid. From Taylor et al (Taylor et al. 2018).
Testing: other
Laboratory testing in patients with untreated Refsum disease should identify elevated serum levels of phytanic acid. If this is found, then genetic confirmation may be sought.
Electromyography with nerve conduction velocities may show segmental demyelination and progressive peripheral sensorimotor axonal loss; autonomic studies may show sudomotor axonal loss (Kuntzer et al. 1993).
Goldman perimetry may show significant constriction of visual fields (Ruether et al. 2010).
Imaging
Imaging findings in Refsum disease are non-specific. Dubois and colleagues (Dubois et al. 1991) describe MRI findings in two cases of IRD (infantile Refsum disease) as follows:
The literature describes a subtle change in the white matter on pathologic specimens obtained for hypomyelination rather than active demyelination. Descriptions of pathological specimens confirm that IRD [infantile Refsum disease] causes a diffuse hypomyelination of the brainstem, the olivary, and the dentate nuclei. Focal loss of myelin sheaths with accumulation of fat-laden macrophages and reactive gliosis are found on pathologic specimens. These findings explain the signal abnormalities of the dentate nuclei. No other abnormalities are detected on MR images. To our knowledge this is the first MR study of IRD in children.
A pattern of symmetrical signal abnormalities on MR images involving the region of the dentate nuclei correlates with some pathologic descriptions and appears to be highly suggestive of IRD. However, it might be a difference in the severity of the affection.
Histopathology
Hallpike (Hallpike 1967) described the case of a male diagnosed with Refsum disease who died at age 38 years. He reported the following temporal bone findings:
Histological examination of the temporal bones revealed normal tympana and labyrinth capsules. Within the membranous labyrinths how-ever, widespread and severe changes had occurred. The changes were very similar in the two labyrinths, both in character and degree. Details of the changes in the left labyrinth are shown in Figs. 5-8.
In […] the cochlea. Reissner’s membrane has collapsed, with obliteration of the scala media. The organ of Cortiis disorganized through-out the cochlea, together with severe degeneration of the stria vascularis. Of the spiral ganglion cells and cochlear nerve fibres, only a few remain.
In […] the organ of Corti and stria vascularis. The cell mass of the organ of Corti has collapsed with retraction of the tectorial membrane into the internal sulcus. The nuclei of the limbus cells are still numerous and well preserved. The degenerate remnants of the stria contain some PAS-positive deposits. Both of the perilymph scalae are lined by a layer of amorphous PAS-positive material.
In […] the saccule. At one point its wall has collapsed, else-where it has ruptured. The sensory epithelium is grossly disorganized and contains aggregations of amorphous PAS-positive material.
In […] the crista of the horizontal canal. It shows no structural abnormality, with good preservation of the sensory epithelium and the sub-epithelial nerve fibres. The sense organs of the utricle and the vertical canals are also well preserved.
Autopsy studies of patients with Refsum disease show involvement of other organ systems as well (Chow et al. 1992).
Differential diagnosis
The differential diagnosis includes disorders such as:
- Several hereditary diseases such as:
- Autoimmune disorders that can potentially involve the vestibulo-cochlear and auditory systems, such as:
Management
The management of Refsum disease is usually multidisciplinary, and depends on the particular clinical and laboratory features that manifest in a given patient.
- The mainstay of therapy for Refsum disease is restriction of dietary phytanic acid intake. Consultation with a medical dietician is appropriate. Acute symptomatic exacerbations are sometimes treated with plasmapheresis to lower serum phytanic acid rapidly (Dickson et al. 1989; Gibberd 1993; Hungerbuhler et al. 1985; Weinstein 1999).
- Consultation with ophthalmology is appropriate to identify and monitor retinitis pigmentosa.
- Consultation with audiology is appropriate to characterize and monitor hearing loss, and to consider amplification when necessary.
- Consultation with physical therapy is reasonable in patients with vestibular weakness and/or cerebellar ataxia.
Prognosis
A diet that strictly limits intake of phytanic acid can retard progression of Refsum disease. For example, Djupesland and colleagues (Djupesland et al. 1983) reported a case of a patient with Refsum disease in whom dietary restriction of phytanic acid stabilized hearing loss, and this was maintained at 15 years. Some findings may improve; for example, vestibular abnormalities in Refsum disease can improve with strict dietary therapy (Taylor et al. 2018), in patients with retinopathy, the electroretinographic responses may improve (Benson et al. 2020).
Additional notes
Refsum disease, while rare, is notable for several features, including:
- It is one of the hereditary causes of “deaf-blindness” (auditory and visual loss).
- It is a potentially treatable cause of multiple deficits (retinopathy, sensorineural hearing loss, vestibular weakness, peripheral neuropathy and cerebellar ataxia) with dietary intervention.
References
Allen IV, Swallow M, Nevin NC, McCormick D (1978) Clinicopathological study of Refsum’s disease with particular reference to fatal complications. J Neurol Neurosurg Psychiatry 41: 323-32. doi: 10.1136/jnnp.41.4.323
Arous S, Atlas I, Arous A, Zahidi H, Benouna EGM, Habbal R (2024) Dilated cardiomyopathy revealing Refsum disease: a case report. J Med Case Rep 18: 470. doi: 10.1186/s13256-024-04789-5
Bamiou DE, Spraggs PR, Gibberd FB, Sidey MC, Luxon LM (2003) Hearing loss in adult Refsum’s disease. Clin Otolaryngol Allied Sci 28: 227-30. doi: 10.1046/j.1365-2273.2003.00694.x
Benson MD, MacDonald IM, Sheehan M, Jain S (2020) Improved electroretinographic responses following dietary intervention in a patient with Refsum disease. JIMD Rep 55: 32-37. doi: 10.1002/jmd2.12147
Bergsmark J, Djupesland G (1968) Heredopathia atactica polyneuritiformis (Refsum’s diseases). An audiological examination of two patients. Eur Neurol 1: 122-30. doi: 10.1159/000113655
Chow CW, Poulos A, Fellenberg AJ, Christodoulou J, Danks DM (1992) Autopsy findings in two siblings with infantile Refsum disease. Acta Neuropathol 83: 190-5. doi: 10.1007/BF00308478
Claridge KG, Gibberd FB, Sidey MC (1992) Refsum disease: the presentation and ophthalmic aspects of Refsum disease in a series of 23 patients. Eye (Lond) 6 ( Pt 4): 371-5. doi: 10.1038/eye.1992.76
Davies MG, Marks R, Dykes PJ, Reynolds D (1977) Epidermal abnormalities in Refsum’s disease. Br J Dermatol 97: 401-6. doi: 10.1111/j.1365-2133.1977.tb14248.x
De Munter S, Verheijden S, Regal L, Baes M (2015) Peroxisomal Disorders: A Review on Cerebellar Pathologies. Brain Pathol 25: 663-78. doi: 10.1111/bpa.12290
Dick AD, Jagger J, McCartney AC (1990) Refsum’s disease: electron microscopy of an iris biopsy. Br J Ophthalmol 74: 370-2. doi: 10.1136/bjo.74.6.370
Dickson N, Mortimer JG, Faed JM, Pollard AC, Styles M, Peart DA (1989) A child with Refsum’s disease: successful treatment with diet and plasma exchange. Dev Med Child Neurol 31: 92-7. doi: 10.1111/j.1469-8749.1989.tb08416.x
Djupesland G, Flottorp G, Refsum S (1983) Phytanic acid storage disease: hearing maintained after 15 years of dietary treatment. Neurology 33: 237-40. doi: 10.1212/wnl.33.2.237
Dotti MT, Rossi A, Rizzuto N, Hayek G, Bardelli N, Bardelli AM, Federico A (1985) Atypical phenotype of Refsum’s disease: clinical, biochemical, neurophysiological and pathological study. Eur Neurol 24: 85-93. doi: 10.1159/000115767
Dubois J, Sebag G, Argyropoulou M, Brunelle F (1991) MR findings in infantile Refsum disease: case report of two family members. AJNR Am J Neuroradiol 12: 1159-60.
Feldmann H (1981) Refsum-Syndrom Heredopathia atactica polyneuritiformis in der Sicht des HNO Arztes*. Laryngo-Rhino-Otol 60: 235-240. doi: 10.1055/s-2007-1008709
Ferdinandusse S, Zomer AW, Komen JC, van den Brink CE, Thanos M, Hamers FP, Wanders RJ, van der Saag PT, Poll-The BT, Brites P (2008) Ataxia with loss of Purkinje cells in a mouse model for Refsum disease. Proc Natl Acad Sci U S A 105: 17712-7. doi: 10.1073/pnas.0806066105
Geisbrecht BV, Collins CS, Reuber BE, Gould SJ (1998) Disruption of a PEX1-PEX6 interaction is the most common cause of the neurologic disorders Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Proc Natl Acad Sci U S A 95: 8630-5. doi: 10.1073/pnas.95.15.8630
Gelot A, Vallat JM, Tabaraud F, Rocchiccioli F (1995) Axonal neuropathy and late detection of Refsum’s disease. Muscle Nerve 18: 667-70. doi: 10.1002/mus.880180617
Gibberd FB (1993) Plasma exchange for Refsum’s disease. Transfus Sci 14: 23-6. doi: 10.1016/0955-3886(93)90049-Z
Gibberd FB, Feher MD, Sidey MC, Wierzbicki AS (2004) Smell testing: an additional tool for identification of adult Refsum’s disease. J Neurol Neurosurg Psychiatry 75: 1334-6. doi: 10.1136/jnnp.2003.026690
Hallpike CS (1967) Observations on the structural basis of two rare varieties of hereditary deafness. In: de Reuck AVS, Knight J (eds) Myotatic, Kinesthetic and Vestibular Mechanisms. CIBA-Foundation, pp 285-294
Harris CM, Shawkat F, Russell-Eggitt I, Wilson J, Taylor D (1996) Intermittent horizontal saccade failure (‘ocular motor apraxia’) in children. Br J Ophthalmol 80: 151-8. doi: 10.1136/bjo.80.2.151
Hungerbuhler JP, Meier C, Rousselle L, Quadri P, Bogousslavsky J (1985) Refsum’s disease: management by diet and plasmapheresis. Eur Neurol 24: 153-9. doi: 10.1159/000115788
Jansen GA, Hogenhout EM, Ferdinandusse S, Waterham HR, Ofman R, Jakobs C, Skjeldal OH, Wanders RJ (2000) Human phytanoyl-CoA hydroxylase: resolution of the gene structure and the molecular basis of Refsum’s disease. Hum Mol Genet 9: 1195-200. doi: 10.1093/hmg/9.8.1195
Jansen GA, Ofman R, Ferdinandusse S, Ijlst L, Muijsers AO, Skjeldal OH, Stokke O, Jakobs C, Besley GT, Wraith JE, Wanders RJ (1997) Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene. Nat Genet 17: 190-3. doi: 10.1038/ng1097-190
Kuntzer T, Ochsner F, Schmid F, Regli F (1993) Quantitative EMG analysis and longitudinal nerve conduction studies in a Refsum’s disease patient. Muscle Nerve 16: 857-63. doi: 10.1002/mus.880160809
Lee ZH, Kim H, Ahn KY, Seo KH, Kim JK, Bae CS, Kim KK (2000) Identification of a brain specific protein that associates with a refsum disease gene product, phytanoyl-CoA alpha-hydroxylase. Brain Res Mol Brain Res 75: 237-47. doi: 10.1016/s0169-328x(99)00304-6
Mihalik SJ, Morrell JC, Kim D, Sacksteder KA, Watkins PA, Gould SJ (1997) Identification of PAHX, a Refsum disease gene. Nat Genet 17: 185-9. doi: 10.1038/ng1097-185
Nyberg-Hansen R (1992) Sigvald Refsum (1907-1991). J Neurol Sci 107: 125-126.
Oysu C, Aslan I, Basaran B, Baserer N (2001) The site of the hearing loss in Refsum’s disease. Int J Pediatr Otorhinolaryngol 61: 129-34. doi: 10.1016/s0165-5876(01)00559-6
Portsteffen H, Beyer A, Becker E, Epplen C, Pawlak A, Kunau WH, Dodt G (1997) Human PEX1 is mutated in complementation group 1 of the peroxisome biogenesis disorders. Nat Genet 17: 449-52. doi: 10.1038/ng1297-449
Refsum S (1946) Heredopathica atactica polyneuritiformis: a familial syndrome not hitherto described. Acta Psychiatrica et Neurologica Scandinavica 38: 1-303.
Refsum S (1952) Heredopathia atactica polyneuritiformis. J Nerv Ment Dis 116: 1046-50. doi: 10.1097/00005053-195212000-00051
Refsum S (1981) Heredopathia atactica polyneuritiformis (phytanic-acid storage disease, Refsum’s disease): a biochemically well-defined disease with a specific dietary treatment. Arch Neurol 38: 605-6. doi: 10.1001/archneur.1981.00510100033003
Refsum S, Salomonsen L, Skatvedt M (1949) Heredopathia atactica polyneuritiformis in children. J Pediatr 35: 335-43. doi: 10.1016/s0022-3476(49)80006-0
Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, Valle D, Gould SJ (1997) Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet 17: 445-8. doi: 10.1038/ng1297-445
Ruether K, Baldwin E, Casteels M, Feher MD, Horn M, Kuranoff S, Leroy BP, Wanders RJ, Wierzbicki AS (2010) Adult Refsum disease: a form of tapetoretinal dystrophy accessible to therapy. Surv Ophthalmol 55: 531-8. doi: 10.1016/j.survophthal.2010.03.007
Salisachs P (1982) Is the “cerebellar” incoordination of Refsum’s disease due to structural lesions in the cerebellum? J Neurol Neurosurg Psychiatry 45: 473-4. doi: 10.1136/jnnp.45.5.473
Taylor RL, Jankelowitz SK, Young AS, Sullivan D, Halmagyi GM, Welgampola MS (2018) Reversible vestibular neuropathy in adult Refsum disease. Neurology 90: 890-892. doi: 10.1212/WNL.0000000000005472
Tran D, Greenhill W, Wilson S (2011) Infantile refsum disease with enamel defects: a case report. Pediatr Dent 33: 266-70.
van den Brink DM, Brites P, Haasjes J, Wierzbicki AS, Mitchell J, Lambert-Hamill M, de Belleroche J, Jansen GA, Waterham HR, Wanders RJ (2003) Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet 72: 471-7. doi: 10.1086/346093
Vandana VP, Bindu PS, Nagappa M, Sinha S, Taly AB (2015) Audiological findings in Infantile Refsum disease. Int J Pediatr Otorhinolaryngol 79: 1366-9. doi: 10.1016/j.ijporl.2015.05.023
Weinstein R (1999) Phytanic acid storage disease (Refsum’s disease): clinical characteristics, pathophysiology and the role of therapeutic apheresis in its management. J Clin Apher 14: 181-4. doi: 10.1002/(sici)1098-1101(1999)14:4<181::aid-jca5>3.0.co;2-z
Weleber RG, Tongue AC, Kennaway NG, Budden SS, Buist NR (1984) Ophthalmic manifestations of infantile phytanic acid storage disease. Arch Ophthalmol 102: 1317-21. doi: 10.1001/archopht.1984.01040031067026
![]()